Примеры из жизни
Под наблюдением Кабинета Фенилкетонурии в детской больнице Кракова в 2009 году находилось 430 пациентов с гиперфенилаланинемией в возрасте от 0 до 56 лет, в том числе 81 женщина старше 16 лет.
Ретроспективно проанализированы все беременности с 1985 по 2009 год
- классической фенилкетонурией (ФА без диетического контроля >20mg/dl)
- умеренной фенилкетонурией (ФА без диетического контроля 10-20mg/dl) и
- Умеренной гиперфенилаланинемией (ФА без диетического контроля 6-10mg/dl) старше 16 лет
Оценке подвергался диетический контроль у женщин с PKU/HPA:
- в пре- и
- постконцепционном периодах
Хорошим признавался контроль, если удавалось достичь:
- стабилизации концентраций ФА в сыворотке крови в границах 2-6mg/dl в преконцепционном периоде и
- сохранить такой уровень контроля в течение всей беременности, a особенно в I и II триместрах
10/50 беременностей наших пациенток были планированы
диета с низким содержанием фенилаланина внедрена 3 месяца перед и ведена на протяжении всей беременности с целью сохранения уровня контроля в границах от 2,0 до 6,0mg/dl
1/10 беременностей закончились самопроизвольным выкидышем
9/10 беременностей закончились рождением здорового ребенка
К сожалению 40/50 были беременностями непланированными: диета не соблюдалась в периоде пре- и посткоцепционном или была введена поздно
8/40 беременностей закончились самопроизвольным выкидышем
- а остальные 32/40 беременностей закончились рождением 24/33 (72,7%) с синдромом материнской ФКУ.
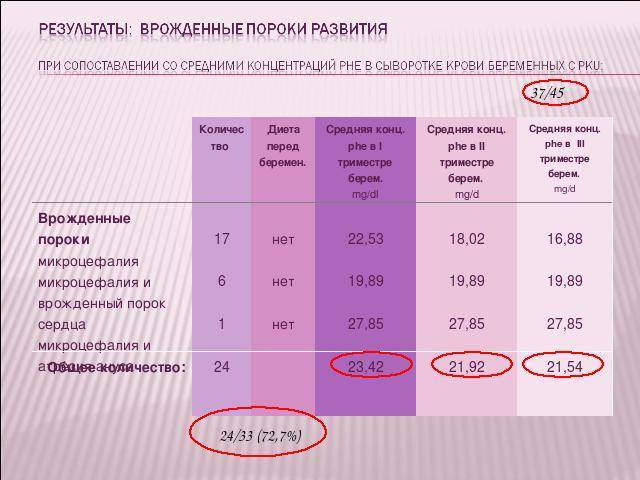
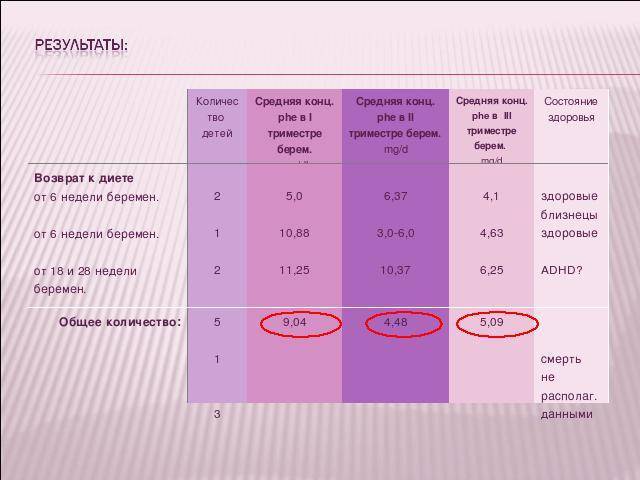
Результаты
Восемь беременных возобновили диету поздно (6-28 нелдели), уровни ФА заранее не контролировались, а при поступлении на учет значительно превышали нормальные. Введденная даже в этом периоде терапия у четырех из десяти пациенток с ФКУ способствовала рождению 5 (15, 79%) клинически здоровых детей.
Пациентка К.К., 17 лет
– диагностировано ФКУ в скрининге новорожденнных
– под опекой Метаболического Центра от момента установления диагноза
– диетическое лечение внедрено в соответствующем периоде, но …
– в подростковом возрасте становилось все более либеральным, контрольные концентрации ФА были очень высокими
– оценка интеллектуального развития на нижней границы возрастой нормы
– недостаточно добросовестное сотрудничество со стороны родителей, отсутствие акцептации болезни окружающими.
Появилась в Клинике на 12 неделе беременности, концетрация ФА, определенная в этом периоде – 20,5мг%. Немедленно при содействии с мужем пациетки внедрено ригористическую диету, что позволило получить метаболический эффект.
K.K. родила дочь без признаков синдрома материнской PKU, но остается под постоянным наблюдением по поводу синдрома ADHD.
Пациентка Б.Г., 30 лет
Сдиагнозирована в периоде новорожденности при помощи пилотажного скрининга, проводимого Институтом Матери и Ребенка в Варшаве до 1985 года. Лечение внедрено от момента установления диагноза – перед окончанием 3 месяца жизни
В нашем наблюдении от 03.08.1993 года
Из анамнеза:
берем. I. самопроизвольный аборт
берем. II, ребенок с синдромом материнской фенилкетонурии – микроцефалией
берем. III, ребенок с синдромом материнской фенилкетонурии – микроцефалией
берем. IV, самопроизвольный аборт
От 1999 года у пациентки наблюдаются невро-вегетативные нарушения, депрессия, в связи с чем остаётся в постоянном неврологическом и психиатрическом лечении
берем. V, явилась на 6 неделе.
Общее описание
Фенилкетонурия (ФКУ) — это тяжкое наследуемое заболевание, происходящее вследствие неправильного обмена ароматических аминокислот и манифестирующееся олигофренией, задержкой физического развития, локомоторными расстройствами. ФКУ заболевает примерно один из 5-10 тыс. новорожденных. Среди больных преобладают девочки, т.к. младенцы мужского пола практически все погибают во время первого года жизни.
В результате того что при ФКУ не расщепляется аминокислота фенилаланин, поступающая с белковой пищей, в организме накапливаются соединения, обладающие нейротоксическим действием, следствием чего является развитие олигофрении. ФКУ наследственно детерминирована и возникает только при условии, что каждый из родителей передал ребенку патологический ген. Практически такое возможно при близкородственных браках. Считается, что порядка 2% популяции являются носителями такого типа гена, но при этом не имеют признаков заболевания.









Ответы на часто задаваемые вопросы
Как проявляется фенилкетонурия у новорожденных?
Как выглядят больные фенилкетонурией?
- посветление волос и радужки глаза из-за недостатка пигмента меланина
- чрезмерная прибавка в весе
- быстро зарастает большой родничок
- суховатая кожа
- шелушение, сыпь и экзема
- частая рвота
- моча и пот с характерным «мышиным» запахом
- появляются судороги и спазмы
- скованность движений и зажатая «поза портного», что связанно с повышенным напряжением в мышцах
- неадекватное поведение, выкрики, смех
- уменьшение размеров черепа
- деформация ушных раковин
- дрожание пальцев рук
- недержание мочи
- выступающая вперед нижняя челюсть
Какие смеси использовать для ребенка с фенилкетонурией?
витаминымикроэлементыДля детей до одного года рекомендуют:
- Афенилак 13, Афенилак 15 от компании “Нутритек”, Россия;
- MIDмил ФКУ 0 (Hero, Испания);
- ХР Аналог (“Нутриция”, Голландия);
- Фенил Фри 1 (“Мид Джонсон” США).
Для детей старше одного года и для взрослых:
- П-АМ 1, П-АМ 2, П-АМ 3;
- Изифен (готовый продукт), а также ХР Максамейд и ХР Максамум с нейтральным и фруктовым вкусами (“Нутриция”, Голландия).
Какие бывают типы фенилкетонурии?
фенилкетонурии
- Фенилкетонурия I. Классическая и наиболее распространенная форма заболевания, описанная выше в статье. Связана с мутацией гена в 12-й хромосоме, при этом нарушается образование фермента фенилаланин-4-гидроксилазы, который превращает фенилаланин в тирозин.
- Фенилкетонурия II. При этой форме заболевания нарушение происходит в 4-й хромосоме. Нарушается выработка фермента дигидроптеридинредуктазы, который также способствует превращению фенилаланина в тирозин. Заболевание наследуется так же, как и I форма: для того, чтобы родился больной ребенок, необходимо, чтобы носителями гена были оба родителя. Распространенность фенилкетонурии II – 1 случай на 100 000 новорожденных.
- Фенилкетонурия III. В результате генетических нарушений возникает недостаток фермента 6-пирувоилтетрагидроптеринсинтазы. Наследуется, как и две предыдущие формы заболевания. Распространенность – 1 случай на 300 000 новорожденных.
Дают ли инвалидность при фенилкетонурии?
Критерии установления инвалидности при фенилкетонурии
- При фенилкетонурии I инвалидность устанавливают только при необратимых нарушениях со стороны центральной нервной системы, которые приводят к неврологическим расстройствам и умственной отсталости.
- При фенилкетонурии II и III типа группу инвалидности устанавливают во всех случаях.
Существует ли профилактика фенилкетонурии?
- Генетическое консультирование. Необходимо людям, планирующим завести ребенка, которые больны или являются носителями неправильного гена, у которых болен хотя бы один близкий родственник или уже родился больной ребенок. Консультирование проводит врач-генетик. Он помогает разобраться, как ген, ответственный за фенилкетонурию, передавался в предыдущих поколениях, каковы риски будущего ребенка. Также генетик помогает с планированием семьи.
- Скрининг новорожденных. Анализ не помогает предотвратить заболевание, но позволяет выявить его максимально рано, пока оно еще не привело к необратимым изменениям в головном мозге.
- Консультации и диета для женщин, страдающих фенилкетонурией. Если вы женщина и страдаете ФКУ, вам следует проконсультироваться с врачом и спросить, когда лучше планировать беременность в вашем случае. Во время беременности нужно соблюдать правильную диету – это помогает предотвратить дефекты развития у ребенка.
Каковы факторы риска фенилкетонурии?
- Как уже упоминалось в статье, ребенок рискует получить заболевание или стать носителем мутантного гена, если он есть у обоих родителей.
- Среди разных этнических групп распространенность фенилкетонурии различается. Например, среди представителей негроидной расы неправильный ген встречается реже.
- В группе повышенного риска находятся дети матерей, страдающих фенилкетонурией. Если во время беременности женщина не придерживается специальной диеты, у ребенка могут возникать дефекты развития.
Симптомы фенилкетонурии
При рождении и в первые недели жизни состояние новорожденного не отличается от нормального. Первые симптомы появляются примерно в 3-месячном возрасте и постепенно усиливаются. К ним относятся:
- торможение психомоторного развития ребенка,
- периодическая рвота, кожная сыпь, припадки,
- ребенок начинает издавать характерный «мышиный запах»,
- повышенный или пониженный мышечный тонус,
- поведенческие расстройства, гиперактивность,
- ослабление концентрации внимания,
- характерная бледная кожа, светлые волосы и бледно-голубые глаза.
 Гиперактивность
Гиперактивность
Фенилкетонурию у младенца можно выявить с помощью генетического теста.
Симптомы нелеченной или поздно пролеченной фенилкетонурии включают следующие проблемы:
- Тяжелая умственная отсталость, часто с практически невозможной оценкой значений IQ, IQ при нелеченой фенилкетонурии обычно не превышает 50.
- Эпилептические припадки, часто очень плохо контролируемые даже при комплексном лечении.
- Нарушения речи, при полной невозможности общения в самых запущенных случаях.
- Нарушения походки — пациенты с наиболее тяжелыми заболеваниями, особенно в пожилом возрасте, передвигаются только с помощью третьих лиц или постоянно неподвижны в постели.
- Неврологические расстройства, прогрессирующие с возрастом, такие как изменение мышечного тонуса, усиление глубоких рефлексов, тремор конечностей, атаксия, спастический пара-или тетрапарез и другие.
- Микроцефалия.
- Экзема, высыпания на коже.
 Тремор конечностей
Тремор конечностей
Наиболее частыми нарушениями поведения при фенилкетонурии являются:
- гиперактивность с неконтролируемыми истериками;
- приступы агрессии по отношению к другим и к себе; последнее может привести к серьезным самоповреждениям;
- деструктивное поведение;
- нарушения сна;
- дефицит внимания и нарушения концентрации внимания;
- аутичное поведение, в т.ч. боязнь незнакомцев и изменения окружающей среды;
- психоз.
Что такое Фенилкетонурия (ФКУ) у детей –
Фенилкетонурия (ФКУ) у детей — генетическая болезнь, которая характеризуется нарушениями обмена фенилаланина и бывает у 1 из 8000–15 000 новорожденных. Форм фенилкетонурии (ФКУ) всего 4, но существует 400 разных мутаций и метаболические фенотипы заболевания.
Фенилкетонурия — наследственная аминоацидопатия, при которой снижается интеллект ребенка, и возникает неврологический дефицит.
Фенилкетонурия I (классическая или тяжелая) – это аутосомно-рецессивное заболевание, которое возникает вследствие мутации гена фенилаланингидроксилазы. В основе заболевания лежит нехватка фенилаланин-4-гидроксилазы которая обеспечивает превращение фенилаланина в тирозин, результатом чего становится накопление фенилаланина и его метаболитов в тканях и физиологических жидкостях организма ребенка.
Отдельную группу представляю атипичные варианты фенилкетонурии. При них симптомы очень похожи на таковые при классическом варианте заболевания. Но нет положительных продвижений по показателям развития ребенка, даже если проводить нужную диетотерапию. Такие варианты объясняются нехваткой дегидроптеринредуктазы, тетрагидроптерина, гуанозин-5-трифосфатциклогидролазы, 6-пирувоилтетрагидроптеринсинтазы и пр.
Фенилкетонурия II (атипичная) — аутосомно-рецессивная болезнь, при которой генный дефект находится в коротком плече хромосомы 4. Характеризуется она нехваткой дегидроптеринредуктазы, что приводит к нарушению восстановления активной формы тетрагидробиоптерина, а в спинномозговой жидкости и сыворотке крови снижается уровень фолатов. Результат таких изменений – метаболические блоки в механизмах превращения фенилаланина в тирозин. Заболевание было выявлено еще в конце 20-го века.
Фенилкетонурия III (атипичная) — аутосомно-рецессивная болезнь, которая вызвана недостаточностью 6-пирувоилтетрагидроптеринсинтазы. Он принимает участие в организме в процессе создания тетрагидробиоптерина из дигидронеоптеринтрифосфата, что было открыто в конце 20-го столетия. Нарушения сходы с таковыми при выше описанной (второй) форме.
Примаптеринурия — атипичная фенилкетонурия у детей с легкой гиперфенилаланинемией, у которых присутствует в больших количества в моче примаптерин и часть его производных, а в спинномозговой жидкости нормальная концентрация нейромедиаторных метаболитов.
Материнская ФКУ – болезнь, при которой снижается уровень интеллекта (вплоть до умственной отсталости) среди потомства женщин, которые больны фенилкетонурией и не сидели на специальной идете, когда были совершеннолетними.
Есть предположения, что при материнской ФКУ нарушения в развитии белого вещества мозга ответственны за формирование неврологического дефицита. Было проведено исследование в 2008 году Кочем и его командой. У младенца, рожденного от матери с ФКУ, при аутопсии головного было найдено некоторое количество патологических изменений: вентикуломегалия, низкий вес мозга, задержка миелинизации (признаков астроцитоза не наблюдалось), гипоплазия белого вещества.
В некоторых странах СНГ применяется условная классификация рассматриваемого заболевания по уровню содержания в сыворотке крови фенилаланина:
Название формы | Уровень фенилаланина |
классическая | выше 20 мг% (1200 мкмоль/л) |
средняя | 10,1–20 мг% (600–1200 мкмоль/л) |
легкая | до 8 мг% (480 мкмоль/л) |
Что делать, если
Тошнота и рвота
– суточная доза безфенилаланиновой формулы разбивать на много маленьких частей
– формула холодная, из холодильника
– частые приемы пищи, небольшие порции, по возможности
– любимые продукты
– между едой сосать маленькие кусочки льда
Запор
– больше жидкостей – негазированная вода, вода с мёдом
– больше целлюлозы (овощи, фрукты, отруби)
– добавка растительных масел к пище
– пробиотики
– пища варeная, не жареная
Госпитализация
– взять с собой формулу, малобелковые продукты (хлеб, макароны, рис и пр.)
– информация медперсонала о заболевании и принципах диетотерапии
– контакт с диетологом или врачом-метаболистом
- Чуство голода
– мелкие закуски между основными блюдами (желательно овощи и фрукты, не сладости)
– следить за прибавкой веса
Характеристика олигоанурической стадии хронической почечной недостаточности
Стадия клинических проявлений, также известная как азотемическая или олигоанурическая, характеризуется специфическими отклонениями, свидетельствующими о значительном повреждении почек.
У больных наблюдается ряд симптомов:
- Изменение объема мочи. Если на первой стадии жидкости выделялось больше нормы, то на второй стадии ХПН объем мочи снижается до 500 мл/сутки или развивается анурия – 50 мл/сутки.
- Признаки интоксикации. Рвота, диарея, тошнота, кожа бледная, сухая, позднее с желтушным оттенком. Из-за повышения мочевины развивается сильный зуд, расчесы не заживают. Больной слабеет, теряет вес, не ест, вплоть до анорексии.
- Специфический запах аммиака изо рта. Из-за нарушения азотистого баланса.
- Почечная отечность. Сначала отекает лицо, затем конечности и туловище.
- Головокружение, головные боли, расстройство памяти.
- Ощущение холода в конечностях. Снижение их чувствительности.
- Двигательные расстройства.
Эти внешние признаки свидетельствуют о наличии сопутствующих состояний, вызванных дисфункцией почек:
- Азотемия. Возникает из-за увеличения продуктов азотистого обмена в крови. Повышается креатинин в плазме. Содержание мочевой кислоты не показательно, так как ее концентрация увеличивается по другим причинам.
- Гиперхлоремический ацидоз. Усиливает гиперкалиемию и гиперкатаболизм. Развивается из-за нарушения механизма всасывания кальция и очень характерен для стадии клинических проявлений. Характерные симптомы: одышка, сильная слабость.
- Гиперкалиемия. Наиболее частый и опасный симптомом ХПН. Почка способна поддерживать функцию всасывания калия вплоть до терминальной стадии, однако гиперкалиемия зависит не только от функционирования органа и при повреждении развивается на начальных стадиях. При чрезмерно высоком содержании калия в плазме – более 7 мэкв/л нервные и мышечные клетки теряют способность к возбудимости, что приводит к параличу, брадикардии, поражению ЦНС, острой дыхательной недостаточности и др. проблемам.
- Спонтанное снижение потребления белка. Связано со снижением аппетита и на фоне интоксикации. Это приводит к гиперкатаболизму и гипоальбуминемии – снижению альбумина в сыворотке крови.
Еще один характерный признак для пациентов с хронической почечной недостаточностью — передозировка препаратов. При ХПН побочные эффекты любого препарата гораздо более выражены, а передозировка возникает в самых неожиданных случаях. Это связано с дисфункцией почки, не способной выводить продукты распада, что приводит к их накоплению в крови.










Диагностика
Чтобы узнать, имеет ли малыш данную патологию, необходимо записаться на прием к врачу-генетику и педиатру. Постановка диагноза осуществляется после выполнения некоторых анализов и осмотра.
Чем раньше будет выявлена болезнь, тем меньше окажется последствий. Именно поэтому кровь для проверки на ФКУ берут еще в роддоме в первую неделю жизни новорожденного. На специальный лист бумаги наносят каплю капиллярной крови спустя час после кормления. При выявлении повышенной концентрация фенилаланина малыша отправляют на консультацию к врачу-генетику в генетический центр.
Если не была выполнена ранняя диагностика заболевания, позже может возникнуть тяжелое физическое и умственное недоразвитие.
История
Фенилкетонурия как болезнь, протекающая с задержкой интеллектуального и психического развития, открыта в 1934 году норвежским ученым-исследователем Иваром Асбьером Феллингом, отсюда и другое обозначение патологии – болезнь Феллинга.
Первых успехов в коррекции здоровья малышей с ФКУ добились медики во главе с Хорстом Биккелем в середине прошлого века. Разработка терапии и ее внедрение проводились на базе Бирмингемского детского госпиталя (Англия).
Однако больших положительных результатов ученым удалось добиться, когда начала широко проводится ранняя диагностика новорожденных на выявление фенилкетонурии, это примерно 1958-1961 год прошлого века.
Расширение возможностей диагностики со временем позволило установить, что в развитии заболевания принимает участие только ген фенилаланингидроксилазы (РАН).
За последние десятилетия описаны атипичные варианты течения болезни, разработаны и широко применяются новейшие способы ее коррекции. В перспективе – использование генотерапии, что вполне вероятно позволит полностью победить болезнь.
Симптомы Фенилкетонурии (ФКУ) у детей:
Новорожденный ребенок не похож на больного. Симптомы фенилкетонурии (ФКУ) начинают быть заметны в возрасте 2-6 месяцев. Типичные проявления:
- отсутствие интереса к окружающему миру
- выраженная вялость
- рвота
- беспокойство
- повышенная раздражительность
С 6 месяцев у малыше заметно отставание в психическом развитии. У меньшинства детей это олигофрения в слабой степени. А более чем у половины детей фиксируют идитию. Рост малыша с ФКУ может быть нормальным или сниженным. Зубки режутся поздно, череп может иметь размеры меньше нормы. Сидеть и ходить ребенок с фенилкетонурией начинает поздно.
Детей с рассматриваемым диагнозом можно отличить по позе и походке. Они широко расставляют ноги, сгибая их в тазобедренном и коленных суставах. Шаги мелкие. При ходьбе ребенок покачивается. Сидят они в так называемом положении портного – поджав ноги, поскольку у них повышен мышечный тонус.
При фенилкетонурии (ФКУ) дети обычно имеют голубой цвет глаз и светлый оттенок волос. Кожа почти не пигментирована. От ребенка слышен «мышиный» запах. В некоторых случаях у больного могут быть припадки эпилепсии, но они проходят по мере взросления ребенка.
Другие типичные симптомы ФКУ у детей:
- дермографизм
- потливость
- повышенная чувствительность к солнечным лучам и травмам
- акроцианоз
- тяжёлая экзема
- дерматит
- склонность к запорам
- артериальная гипотония
- расстройства аутистического спектра
- гиперактивность
Если не провести вовремя лечение, уровень интеллекта ребенка будет составлять менее 50. В возрасте 18 месяцев могут появиться судорожные приступы. Исчезают они спонтанно. В раннем возрасте приступы часто проходят в форме инфантильных спазмов, далее становятся тоникоклоническими припадками.
Патогенез фенилкетонурии
Дефицит фермента фенилалаиин-4-гидроксилазы лежит в основе классической формы заболевания, он приводит к нарушению окисления поступающего с пищей фенилаланина. Его концентрация в крови и спинномозговой жидкости значительно возрастает, а уровень тирозина падает. Это приводит к нарушению миелинизации нервных волокон, снижению образования нейромедиаторов и запускает патогенетические механизмы задержки умственного развития и вызывает прогредиентное слабоумие.
Симптоматика фенилкетонурии
Манифестация болезни Феллинга происходит в возрасте 2–6 мес. Развиваются первые неспецифические симптомы: вялость, беспокойство и гипервозбудимость, мышечная дистония, срыгивание, судорожный синдром. Патогномоничным симптомом фенилкетонурии считается упорная рвота.
После 6 мес. наблюдается отставание ребенка в психомоторном развитии. Это проявляется как снижение активности, безучастность к окружающему, ребенок перестает узнавать родных, не делает попыток сесть или встать на ноги. Могут появляться шелушение кожи, экзема, дерматит, склеродермия.
Дети с фенилкетонурией, не получающие лечения, страдают от микроцефалии, прогнатии, позднего прорезывания зубов, гипоплазии эмали. Для них характерна задержка речевого развития, к трем–четырем годам развивается олигофрения с практически полным отсутствием речи.
Больные имеют диспластическое телосложение, отличаются светлой кожей, волосами и глазами. Верхние и нижние конечности согнуты в суставах (так называемая «поза портного»), походка шаткая, семенящая, может быть тремор рук, гиперкинезы.
Диагностика Фенилкетонурии (ФКУ) у детей:
Для диагностики фенилкетонурии (ФКУ) у детей определяют содержание крови уровней фенилаланина и тирозина в крови. Применяют тест Гатри, пробу Феллинга, флуориметрию, хроматографию, МРТ, поиск мутантного гена, электроэнцефалографию.
ЭЭГ позволяет обнаружить нарушения в основном в виде паттерна гипсартимии, даже если приступов у ребенка не наблюдалось. Также находят фокусы спайк- и полиспайк-разрядов (единичные и множественные). МРТ не находит изменений сигнала в стволе, мозжечке или коре головного мозга. Изменения на МРТ не коррелируют с уровнем интеллекта, они зависят от содержания фенилаланина в крови.
Если у ребенка фенилкетонурия II, то симптомы проявляются после 12 месяцев жизни. В крови затем находят повышенный уровень фенилаланина в периоде новорожденности, назначают диету, но болезнь всё равно прогрессирует. У малышей выраженная умственная отсталость, судороги, признаки повышенной возбудимости, гиперрефлексия, мышечная дистония, спастический тетрапарез. Летальный исход в части случаев наступает в возрасте от 2 до 3 лет.
Симптомы фенилкетонурии III напоминают выше перечисленные. У ребенка врачи обнаруживают три типичных признака:
- спастический тетрапарез
- микроцефалия
- глубокая умственная отсталость
Принципы диагностики
В настоящий момент на территории Росси массово в роддомах проводится неонатальный скрининг – определение наследственных заболеваний, в том числе и ФКУ, на 3-4 день жизни новорожденного.
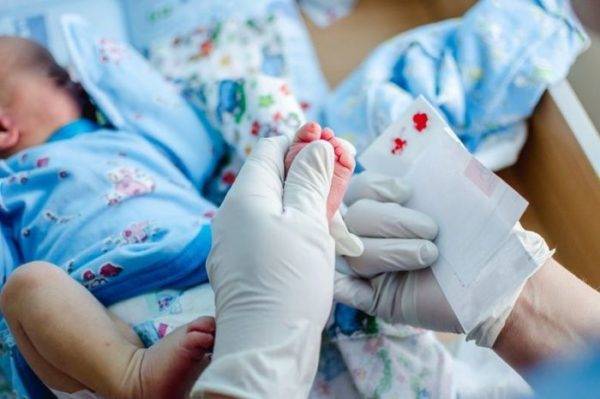
Массовое обследование детей позволяет своевременно выявить фенилкетонурию и назначить соответствующее лечение, что при полном соблюдении терапии исключает дальнейшее прогрессирование патологии.
Аллергия у грудничков, как выглядит, симптомы, причины, какие лекарства можно и что исключить из рациона
В случае отсутствия лечения (но не ранее 10 дня жизни новорожденного) в моче выявляются фенилкетоны – продукты расщепления фенилаланина.
Если в крови при проведении неонатального скрининга обнаруживаются данные за развитие патологии, то дальнейшее обследование должно проводиться на базе медико-генетических центров.
Для подтверждения диагноза необходимы следующие исследования:
- Выявление уровня фенилаланина в сыворотке и высохшем пятне крови;
- Определение количества тирозина;
- Копрограмма;
- Потовый тест;
- ДНК-диагностика;
- Хроматография;
- Проба Феллинга – определение в моче фенилпировиноградной кислоты;
- Электроэнцефалография.
В ряде случаев назначается определение показателей фенилаланингидроксилазы в биоптате печени.
Список использованой литературы:
- С.Б. Гридина, Физиолого-биохимические основы разработки продуктов детского и функционального питания. Учебное пособие, Кемерово, 2004 г.
- В.Г. Кисляковская, Л.П. Васильева, Д.Б. Гурвич “Питание детей раннего и дошкольного возраста”. Пособие для воспитателя детского сада. Москва, Просвещение, 1983 г.
- А.С. Алексеева, Л.В. Дружинина, К.С. Ладодо “Организация питания детей в дошкольных учреждениях”, Пособие для воспитателя детского сада, Москва, Просвещение, 1990 г
- “Методические рекомендации”, Питание детей в детских дошкольных учреждениях, Методические рекомендации по питанию детей в детских дошкольных учреждениях разработаны в Институте питания АМН СССР сотрудниками отдела детского питания проф. Ладодо К.С., проф. Фатеевой Е.М., к.м.н. Кистеневой Г.С., к.м.н. Белкиной Л.М., врачом Степановой Т.Н., диетсестрой Басовой Л.И. и сотрудником отдела химии и технологии пищевых продуктов ст. инженером Чумаковой В.В.
- Санитарные правила устройства и содержания детских дошкольных учреждений (детские ясли, детские сады, детские сады-ясли) утвержденные 20.03.1985 г № 3231-85 г.
- “Кормление и питание грудных детей и детей раннего возраста”. Методические рекомендации для Европейского региона ВОЗ с особым акцентом на республики бывшего Советского Союза, № 87, Unisef, ВОЗ, 2003 г.
- “Организация питания в детских учреждениях”. В.Ф. Ведрашко, Москва, Медицина, 1969 г.
- “Инструкция по организации питания детей в дошкольных учебных заведениях”. Утверждена приказом Минобразовния и науки Украины, МОЗ Украины 17.04.2006 г № 298/227
- К.С. Ладодо, Л.В. Дружинина, А.С. Вынту “Рациональное питание детей раннего возраста”. Кишинев, “Картя Молдавеияскэ”, 1986 г.
- А.М. Гринкевич, Г.Ю. Лазарева, О.И. Чапова “Детское питание”. Изд. дом “Равновесие”, электронная книга.
- М.И. Снигур, З.Т. Корешкова “Питание детей”. Киев, “Радянська школа”, 1988 г.
- Л.Н. Цветкова, В.С. Салмова, Е.Е. Вартапетова “Питание детей старше года”. Методическое пособие для преподавателей, студентов, клинических ординаторов, интернов, практических врачей-педиатров, Кафедра пропедевтики детских болезней РГМУ
- И.М. Скурихин, М.Н. Волгарев “Химический состав пищевых продуктов” Справочник, Москва, “Агропромиздат”, 1987 г.
- «Nährwerttabelle für die Ernährung bei angeborenen Störungen des Aminosäurestoffwechsels» (Пищевая ценность продуктов для питания с врожденными нарушениями метаболизма аминокислот). Методическое пособие, Германия.
- Методические рекомендации “Диагностика, диетотерапия и медико-генетическая консультация при фенилкетонурии у детей”. Киев, 2000 г
- “Організація харчування дітей в дошкільних закладах”. Міжвідомча інструкція.-(Міністерство охорони здоров“я України, Міністерство освіти України) – Київ.- 1993.- 68с.
- “Організація харчування дітей в дошкільних закладах”. Н.В. Омеляненко, Н.М. Курочка, Київ, «Шкiльний свiт», 2010 р. – 160с
Виды
Заболевание может протекать по классическому и атипичному варианту. К атипичным формам болезни относят:
- ФЕНИЛКЕТОНУРИЮ II ТИПА. Мутация гена приводит к дефициту дигидробиоптерин-редуктазы, в результате активность соединения, ответственного за преобразование фенилаланина, нарушается. Одновременно в сыворотке крови и в цереброспинальной жидкости больного выявляется дефицит витамина В9, от нормального количества которого зависит утилизация (расщепление и выведение) аминокислот.
- ФЕНИЛКЕТОНУРИЮ III ТИПА. Развивается вследствие нехватки катализатора, необходимого для выработки тетрагидробиоптерина (преобразует поступающий в организм фенилаланин в тирозин).
- ПРИМАПТЕРИНУРИЮ. Выявляется при незначительной степени гиперфенилаланинемии. До настоящего времени ферментная мутация данного вида фенилкетонурии еще не установлена. Но у больных в моче обнаруживается избыточное количество примаптерина и его соединений, а в спинномозговой жидкости уровень нейромедиаторных метаболитов остается в пределах нормы.
Атипичные типы ФКУ сходны по клиническим симптомам с классическим течением патологии, но при их развитии даже своевременное лечение и диетотерапия не останавливают необратимые изменения в функционировании органов ЦНС.
Отдельно выделяют материнскую фенилкетонурию, заболевание диагностируется у детей, рожденных женщинами с ФКУ, которые не соблюдали лечебное питание.
Повышенный уровень фенилаланина вызывает ряд врожденных аномалий развития головного мозга:
- Вентрикуломегалию – увеличение желудочков головного мозга в размерах;
- Задержку миелинизации (формирование оболочек нервных клеток) и гипоплазию (недостаточное развитие) белого вещества мозга;
- Низкий вес головного мозга по сравнению с нормой.
Материнская фенилкетонурия становится причиной хронической интоксикации развивающегося плода токсическими соединениями, и как следствие этого приводит к задержке развития умственной функции рожденных детей.
Группы продуктов при ФКУ
- Красный список – продукты, которые необходимо полностью исключить из рациона.
- Оранжевый список – разрешены в небольших количествах под строгим контролем.
- Зеленый список – могут употребляться без ограничений.
| Красный список | Оранжевый список | Зеленый список |
| Все виды мяса | Молочные продукты | Фрукты |
| Колбасные изделия | Рис и кукуруза | Ягоды |
| Все виды рыбы | Овощи (картофель, капуста) | Зелень |
| Морепродукты | Овощные консервы | Овощи |
| Яйца | Рисовая, кукурузная мука | |
| Сыры | Крахмал и саго | |
| Творог | Сахар и варенье | |
| Орехи | Мед | |
| Хлеб и хлебобулочные изделия | Сливочное и растительное масло, топленый жир | |
| Кондитерские изделия | ||
| Крупы и хлопья | ||
| Продукты из сои | ||
| Поп-корн | ||
| Аспартам |
- искусственные низкобелковые продукты, специально для диетического питания (хлеб, печенье, макароны)
- готовые пюре для детского питания на основе фруктов.