

Признаки Синдрома Эдвардса
- Низкий вес при рождении независимо от срока беременности;
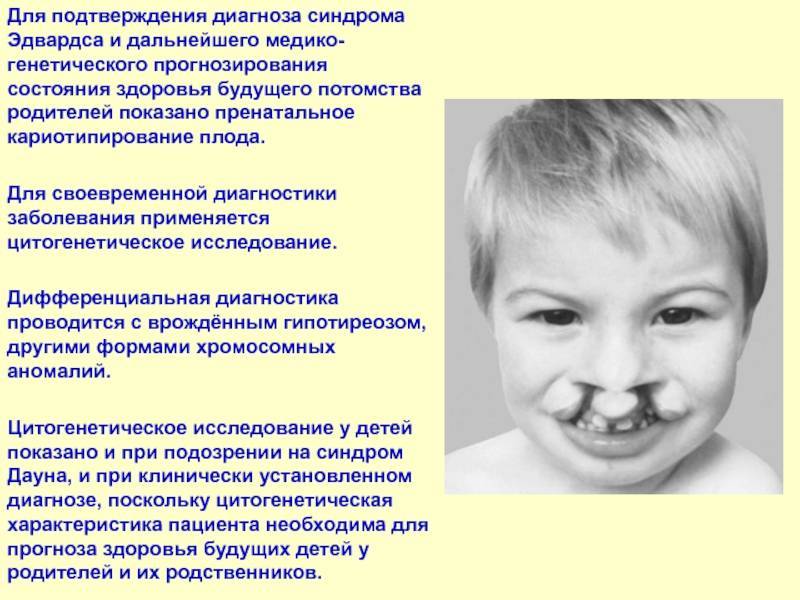
- Характерные изменения головы: деформированный маленький череп, маленький лоб и рот, суженные глаза, неправильной формы уши и др.;
- Микрогнатия (дефекты верхней и нижней челюстей);
- Форма лица искажена, сформирован неправильный привкус;
- «Волчья пасть» и «заячья губа». Расщелины встречаются не у всех детей;
- Множественные пороки развития сердечно-сосудистой, мочевыводящей и пищеварительной систем;
- Нарушение рефлекса сосания и глотания (возникают большие сложности с кормлением);
- Патологии половых органов (у девочек — увеличение клитора, у мальчиков – аномалии полового члена, дефекты расположения яичек);
- Пороки развития опорно-двигательного аппарата (косолапость, сращение пальцев на ногах);
- Практически полное отсутствие физического и умственного развития.
Как часто встречается эта патология?
Дети с синдромом Эдвардса погибают на этапе внутриутробного развития приблизительно в 60% случаев. Несмотря на это, среди генетических заболеваний данный синдром у выживших младенцев является достаточно распространенным, по частоте встречаемости уступая только синдрому Дауна. Распространенность синдрома Эдвардса составляет 1 случай на 3 — 8 тысяч детей.
Смертность при синдроме Эдвардса на первом году жизни составляет около 90%, причем средняя продолжительность жизни при тяжелом течении заболевания у мальчиков — 2–3 месяца, а девочек — 10 месяцев, а до взрослого состояния доживают лишь единицы. Чаще всего дети с синдромом Эдвардса умирают от удушья, пневмонии, сердечно–сосудистой недостаточности или кишечной непроходимости — осложнений, вызванных врожденными пороками развития.
Можно ли избежать возникновения хромосомных аномалий
Что же можно сделать, чтобы избежать анеуплоидии? К сожалению, практически ничего. Анеуплоидия возникает спонтанно во время формирования репродуктивных клеток (сперматазоидов и яйцеклеток) у родителей или при раннем делении клеток во время развития эмбриона. Нет данных о влиянии на этот процесс каких-либо факторов: образа жизни родителей или их состояния здоровья, экологии и т.п. Однако есть данные, указывающие на то, что возраст матери имеет значение в случае таких трисомий, как 21, 18 и 13 (синдромы Дауна, Эдвардса и Патау соответственно). Общая вероятность возникновения одной из трёх этих трисомий к 45 годам беременной повышается в 12,5 раз.
Единственная возможность предотвратить развитие эмбриона с хромосомной аномалией — выполнить его генетический скрининг, и сделать это можно только в рамках протокола ЭКО. Лаборатория Genetico предоставляет такое тестирование — преимплантационное генетическое тестирование на хромосомные аномалии (ПГТ-А).
ПГТ-лаборатория Genetico была открыта в 2014 г. совместно с пионером и мировым лидером в данной области — Институтом Репродуктивной Генетики США.
Этот метод позволяет отобрать для подсадки эмбрионы без хромосомных нарушений. Благодаря этому повышается результативность ЭКО, снижается вероятность рождения ребенка с хромосомными аномалиями, а также риск спонтанного прерывании беременности.
ПГТ-А Эмбриотест
Генетический скрининг хромосомных аномалий, с целью повышения эффективности лечения бесплодия в протоколе ЭКО.
Наши программы пренатального скрининга
В клинике «МедикСити» используются программы международного образца, благодаря которым на ранней стадии и в сжатые сроки выявляется большинство неизлечимых генетических пороков (синдромы Дауна, Патау, Эдвардса, Корнелии де Ланге, Шерешневского-Тернера, патологии развития нервной трубки и др.).
Пренатальный скрининг:
- Пренатальный скрининг I триместра беременности, расчет риска хромосомных аномалий плода, программа LifeCycle (DELFIA)
- РАРР-А
- Свободный b-ХГЧ
- Пренатальный скрининг II триместра беременности, расчет риска хромосомных аномалий плода, программа LifeCycle (DELFIA)
- АФП
- Свободный b-ХГЧ
- Свободный эстриол
- Пренатальный скрининг I триместра с расчетом риска преэклампсии, программы LifeCycle, Predictor (DELFIA)
- PAPP-A
- Свободный b-ХГЧ
- PLGF
- Пренатальный скрининг I триместра беременности, расчет риска хромосомных аномалий плода, программа PRISCA (IMMULITE)
- PAPP-A
- Свободный b-ХГЧ
- Расчет риска ранней и поздней преэклампсии I триместра беременности, программа Predictor (DELFIA)
- PAPP-A
- PLGF
- Пренатальный биохимический скрининг I триместра беременности, без расчета риска (для внесения в программу Astraia).
- Определение биохимических маркеров РАРР-А, свободный b-hCG беременным на сроке от 9 недель до 13 недель 6 дней. Исследование проводится на анализаторе Delfia (PerkinElmer, США) в соответствии с рекомендациями FMF (Fetal Medicine Foundation – Фонд Медицины Плода, Лондон).
Симптомы
Проявления болезни делят на несколько групп. В первую причисляют те, что характеризуют внешний вид больного человека:
- вес тела при рождении составляет примерно 2кг 100 грамм или 2 кг 200 грамм
- ненормально развитая нижняя или верхняя челюсть
- голова маленькая по отношению ко всему телу
- расщелина верхней губы и/или твердого неба
- неправильный прикус и неправильная форма лица ребенка
- стопа-качалка
- косолапость от рождения
- перепонки на пальцах ног или полное слияние пальцев
- уши низко посажены
- пальцы кисти сжаты, наблюдается неровное их расположение в кулачке
- ротовая щель меньше, чем должна быть
Вторая группа симптомов болезни касается нервно-психической сферы, моторики и функции органов больного ребенка:
- пупочная или паховая грыжа
- врожденные пороки сердца, включая открытый артериальный проток, дефект межжелудочковой перегородки и т.п.
- сглаживание или атрофия мозговых извилин
- недоразвитость мозжечка, мозолистого тела
- умственная отсталость
- задержка нервно–психического развития ребенка
- нарушение локации кишечника
- меккелев дивертикул
- атрезия пищевода или заднего прохода
- нарушение глотательного и сосательного рефлекса
- ГЭРБ
- удвоение мочеточников
- подковообразная или сегментированная почка
- недоразвитость яичников у девочек
- гипертрофированный клитор у младенцев женского пола
- гипоспадия у младенцев мужского пола
- крипторхизм у больных мальчиков
- атрофия мышц
- сколиоз
- косоглазие
Лучшие специалисты в Санкт-Петербурге с рейтингом 4.5+
Климанова Дарья Александровна
Специализация: Педиатр, Неонатолог, Аллерголог, Иммунолог
Врачебный стаж: с 2013 года
Где ведет прием: МЦ Балтмед Озерки
Записаться

Виноградова Галина Васильевна
Специализация: Неонатолог
Врачебный стаж: с 1998 года
Где ведет прием: Клиника Скандинавия
Записаться
Литература
- Стрижаков А.И. т Бунин АХ , Медведев M B. Ультразвуковая диагностика в акушерской клинике. М.: Медицина, 1990.
- Роберо Рт Пилу Дж., Дженти Ф., Гили ни Л., Хоббинс. Дж. С- Пренатальная диагностика врожденных пороков развития плода. М.: Медицина, 1994.
- Ворсанова С.Г., Юров Ю.Б., Чернышов В.Н. Хромосомные синдромы и аномалии. Классификация и номенклатура. // Ростов-на-Дону. Молот. – 1999. -192с.
- Алтынник H.A. Значение ультразвуковой оценки толщины воротникового пространства плода в ранние сроки беременности для пренатальной диагностики хромосомных аномалий: Дисс. . канд. мед. наук. М., 2002. – 105 с.
- Снайдерс Р.Дж.М., Николаидес К.Х. Ультразвуковые маркеры хромосомных дефектов плода. М.: Видар, 1997. 175 с.
Синдром Клайнфельтера
Синдром Клайнфельтера – это генетическое заболевание, которое возникает, когда мальчик рождается с лишней копией Х-хромосомы. Синдром часто не диагностируют до взрослого возраста. Синдром Клайнфельтера может негативно повлиять на рост яичек, в результате чего они становятся меньше нормальных. Это приводит к снижению выработки тестостерона.
Читать далее
Синдром Ди Георга
Синдром Ди Георга, более точно известный под более широким термином – синдромом делеции 22q11.2 — это расстройство, вызванное отсутствием небольшого фрагмента хромосомы 22. Состояние приводит к плохому развитию нескольких систем организма.
Читать далее
Как же быть в данном случае?
Благодаря современным научным разработкам появилась альтернатива. Это прямой неинвазивный метод диагностики хромосомных аномалий. Разница заключается в том, что материал для исследования получают не из клеток плода, а из крови матери.
Кровь забирается из локтевой вены как при обычном биохимическом анализе. Диагностику можно проводить на любом сроке беременности начиная с 10 нед. Срок ожидания результата 12 дней








Достоверность метода более 99% (акцентирую внимание – речь идет о диагностике хромосомных аномалий, морфологические нарушения эмбриогенеза мы можем увидеть только на УЗИ)
К сожалению, пока данные анализы приходится проводить в лабораториях США, поэтому стоимость их достаточно высока, но в скором времени они будут выполняться в России и, надеюсь, станут более доступны для наших сограждан.
Что после рождения?
Младенцы, рожденные с синдромом Эдвардса, обычно рождаются недоношенными, с очень низкой массой тела при рождении и требуют интенсивной терапии с самого рождения. Дефекты, связанные с возникновением этого заболевания, могут включать многие системы и органы, в том числе: пороки сердца, гортань, диафрагмальную грыжу, стриктуру пищевода, гидронефроз и грыжу позвоночника.
Выживаемость новорожденных с синдромом Эдвардса составляет около 50% в первые недели жизни. Однако до 90% детей умирают в возрасте до 1 года.
Обычно кормление ребенка необходимо через зонд, введенный в желудок или энтерально, из-за нарушения сосательного и глотательного рефлексов. Малыши также находятся под наблюдением кардиолога, невролога, нефролога, генетика, а также нуждаются в реабилитации для улучшения работы органов движения.
Хромосомная патология – синдром Клайнфельтера
Риск хромосомной патологии плода – синдрома Клайнфельтера, составляет 1 к 600 в среднем. Это мальчики, которые имеют в дальнейшем высокий рост, телосложение по женскому типу, гинекомастию в 100% случаев заболевания. Кариотип патологии – 47. ХХY, 48. ХХХY; 47. ХYY; 48. ХYYY; 49. ХХХYY; 49. ХХХХY.
Люди с такой патологией подвержены внушаемости, эмоциональной лабильности. У них длинные руки, пальцы рук, в 100% случаев микроорхидизм, в период полового созревания появляются яркие признаки патологии – практически отсутствует оволосение в области половых органов, гиалиноз семенных канатиков и дегенерация эпителия, бесплодие. Больные апатичны, безынициативны, склонны к депрессивным психозам, к алкоголизму, асоциальному поведению в обществе. В детстве больные астеничны, взрослые страдают повышенной массой тела.
Больные с синдромом Клайнфельтера и полисомией 47. ХYY, могут выглядеть абсолютно здоровыми людьми, большая часть больных имеет умственное развитие близкое к норме или слегка сниженное. Некоторые больные отличаются агрессивным поведением, имеют хорошее телосложение, развитую мускулатуру, у них высокий рост. Отмечено, что среди преступников-рецидивистов часто встречаются больные с полисомией такого типа.
Бесплатный прием репродуктолога
по 31 января 2023Осталось 17 дней
Уважаемые пациенты! Клиника «Центр ЭКО» приглашает вас на бесплатный прием репродуктолога с проведением УЗИ и составлением плана лечения.
Пример расчета точности НИПТ:
Приведу пример для наглядности: при частоте синдрома Дауна 1/600 из 100 000 беременностей можно ожидать 167 случаев рождения ребенка с болезнью Дауна и 99 833 рождения здоровых детей. При проведении стандартного скрининга среди этих 100 000 беременных женщин мы получим примерно 3 293 ложноположительных теста, которые потребуют ненужных дальнейших инвазивных процедур и создадут стрессовую ситуацию для женщин. Наряду с этим, мы сможем выявить лишь 142 случая синдрома Дауна из 167, то есть 25 случаев останутся нераспознанными на дородовом этапе.При применении НИПТ у этих же 100 000 беременных мы получим лишь 80 ложноположительных тестов, когда потребуется дальнейшая инвазивная диагностика. Выявлено будет 165 случаев синдрома Дауна из 167, то есть упущено будет не более 2-х случаев.
Разница очевидна.
Выбор, безусловно, за вами. Но он должен быть информированным. Проведение НИПТ не является обязательным, однако, современная медицина дает нам такую возможность и мы должны знать о ней, чтобы принять верное для себя решение и не жалеть об упущенных возможностях в дальнейшем. Посоветуйтесь со своим лечащим врачом и примите правильное решение в каждом конкретном случае.
Частота
В 60 случаях мутаций из 100 дети с рассматриваемым синдромом погибают внутри живота матери, потому что из пороки несовместимы с жизнью. Но выживаемость детей с синдромом Эдвардса достаточно высокая (чуть ниже, чем у плодов с трисомией 21). На 3-8 тысяч младенцев один рождается в рассматриваемым диагнозом.
Врачи говорят, что среди младенцев женского пола болезнь встречается в три раза чаще, чем среди мальчиков. Большой риск родить ребенка с данными отклонениями у рожениц, которым более 30 лет. На проятжении первых 12 месяцев жизни умирает около 90 детей из 100 с таким диагнозом. Мальчики живут в среднем от 2 до 3 месяцев, а девочки около 10 месяцев. Шансы, что ребенок с синдромом Эдвардса сможет дожить до взрослых лет, мизерные. Осложнения пороков развития становятся причинами смерти детей:
- кишечная непроходимость
- сердечно–сосудистая недостаточность
- пневмония
- удушье
Лечение
Почти всегда приобретенные при формировании организма пороки являются несовместимыми с жизнью, поэтому лечение сводится к тому, чтобы облегчить симптоматику, продлить жизнь и максимально нормализовать физиологические функции.
Дети, страдающие синдромом Эдвардса, довольно часто болеют синуситом, конъюнктивитом, средним отитом и инфекционными процессами в мочеполовой системе. В связи с этим основным врачом, который обеспечивает поддержание их жизни, является педиатр. Он регулярно проводит осмотры, дает рекомендации по правильному уходу, следит за достаточным питанием.
Хирургическое исправление отклонений слишком опасно и неоправданно, поэтому проводится крайне редко.
О механизме наследования
Большинство случаев синдрома не наследуются. Он возникает из-за случайных событий во время образования яйцеклетки и спермы. В результате ошибки в делении возникает репродуктивная клетка с неправильным числом хромосом. Например, у сперматозоида или яйцеклетки может появиться добавочная копия хромосомы 18. Когда одна из таких нетипичных репродуктивных клеток вносит вклад в генетический состав ребёнка, у него во всех клетках будет дополнительная хромосома 18.
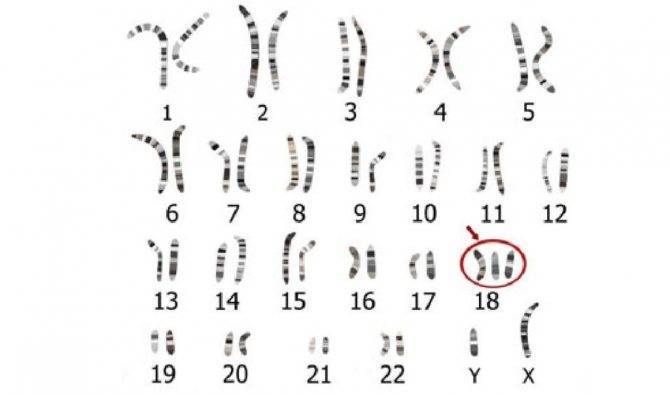
Также не наследуется мозаичная трисомия 18. Она обусловлена случайными событиями при делении клеток в начале эмбрионального развития. В итоге отдельные клетки содержат две копии хромосомы 18, а другие — три копии этой хромосомы.










Может быть унаследована транслокационная трисомия. Незатронутый человек несёт перегруппировку генетического материала между хромосомой 18 и другой хромосомой. Хотя у него нет признаков трисомии 18, человек, у которого есть этот тип транслокации, подвергаются повышенному риску иметь детей с этим генетическим расстройством.
Какова точность теста НИПТ?
НИПТ является высокоточным методом. Его точность по результатам многочисленных исследований составляет до 99,9%. При этом отмечен низкий процент необходимости перезабора крови в связи с недостатком плодовой ДНК в образце (менее 3% случаев), низкий процент ложно положительных результатов (то есть тех случаев, когда при положительном результате теста ребенок оказывается здоров – не более 0,72% или 1 случая на 140). Это означает, что лишь в 1 случае из 140 положительных результатов теста патология плода не будет подтверждена, а в 139 случаях – тест позволит ее выявить.
Частота ложноотрицательных результатов (то есть случаев, когда тест будет отрицательным при имеющейся патологии плода) составляет не более 1 случая на 10 000 тестов.
Тем не менее, принимая решение о проведении теста, нужно полностью отдавать себе отчет в том, что данный тест не является диагнозом, а лишь с очень высокой вероятностью позволяет заподозрить хромосомную патологию плода. В случае если будет заподозрена серьезная патология плода и будет обсуждаться вопрос о возможности/ невозможности сохранения беременности – потребуется подтверждение с помощью дополнительных тестов – биопсии ворсин хориона, амниоцентеза и пр., т.е. инвазивной диагностики. Однако, в связи с крайне низким процентом ложноположительных результатов такие исследования потребуются на порядок реже, чем при проведении стандартного скрининга.
О механизме наследования
Большинство случаев синдрома не наследуются. Он возникает из-за случайных событий во время образования яйцеклетки и спермы. В результате ошибки в делении возникает репродуктивная клетка с неправильным числом хромосом. Например, у сперматозоида или яйцеклетки может появиться добавочная копия хромосомы 18. Когда одна из таких нетипичных репродуктивных клеток вносит вклад в генетический состав ребёнка, у него во всех клетках будет дополнительная хромосома 18.
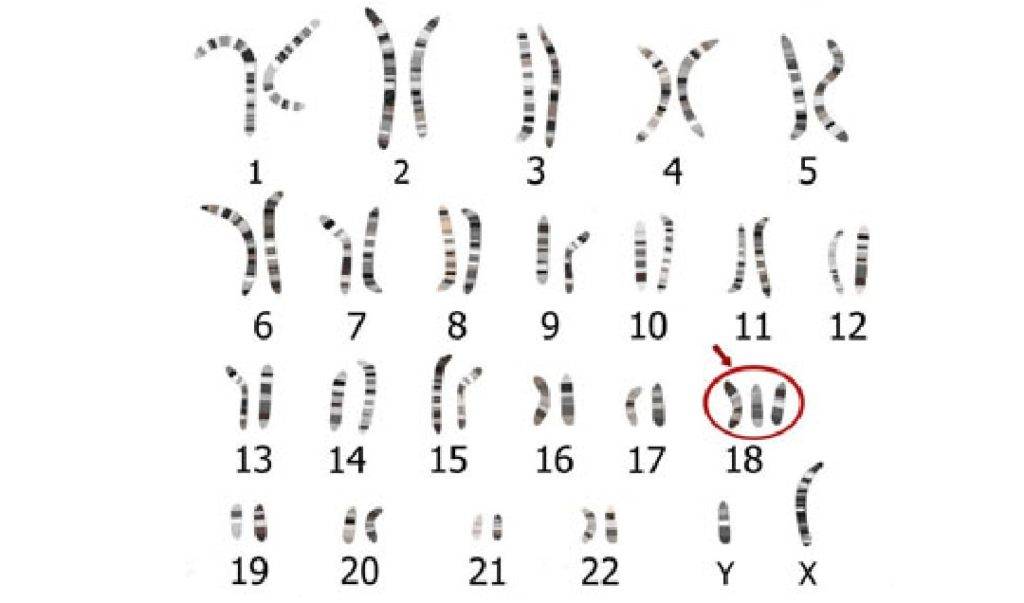
Также не наследуется мозаичная трисомия 18. Она обусловлена случайными событиями при делении клеток в начале эмбрионального развития. В итоге отдельные клетки содержат две копии хромосомы 18, а другие — три копии этой хромосомы.
Может быть унаследована транслокационная трисомия. Незатронутый человек несёт перегруппировку генетического материала между хромосомой 18 и другой хромосомой. Хотя у него нет признаков трисомии 18, человек, у которого есть этот тип транслокации, подвергаются повышенному риску иметь детей с этим генетическим расстройством.
Как узнать патологию во время беременности — диагностика
Синдром Патау, Эдвардса и другие трисомии лучше всего выявить до рождения малыша. Как правило, пренатальная диагностика данного синдрома проводится в 2 этапа:
- На сроке в 11–13 недель (скрининг, в основе которого — проведение различных биохимических анализов у женщины).
- Определение кариотипа плода у беременных их группы риска.
В 11–13 недель в крови женщины определяется уровень некоторых белков крови: β–ХГЧ (β–субъединица хорионического гормона человека) и плазменного протеина А ассоциированного с беременностью. Затем с учетом этих данных, возраста беременной женщины рассчитывается риск рождения ребенка с синдромом Эдвардса, и формируется группа риска беременных.
Далее в группе риска на более позднем сроке берется материал у плода для постановки точного диагноза: в 8–12 недель это биопсия ворсин хориона, в 14–18 — амниоцентез (изучение околоплодных вод), спустя 20 недель — кордоцентез (внутриутробное взятие крови из пуповины плода с УЗИ–контролем). После этого в полученном материале определяют наличие или отсутствие дополнительной 18–й хромосомы с помощью КФ–ПЦР (количественной флуоресцентной полимеразной цепной реакции).
Если беременная не прошла генетическое скрининг–обследование, то на более поздних сроках предварительная диагностика синдрома Эдвардса осуществляется с помощью УЗИ. Прочие косвенные признаки, на основании которых можно заподозрить синдром Эдвардса на более поздних сроках:
- Наличие аномалий развития костей и мягких тканей головы («волчья пасть», микроцефалия, низкая посадка ушных раковин, «заячья губа» и т.п.).
- Обнаружение пороков со стороны сердечно–сосудистой , мочеполовой системы, а также опорно–двигательного аппарата.
Как же быть в данном случае?
Благодаря современным научным разработкам появилась альтернатива. Это прямой неинвазивный метод диагностики хромосомных аномалий. Разница заключается в том, что материал для исследования получают не из клеток плода, а из крови матери.
Кровь забирается из локтевой вены как при обычном биохимическом анализе. Диагностику можно проводить на любом сроке беременности начиная с 10 нед. Срок ожидания результата 12 дней
Достоверность метода более 99% (акцентирую внимание – речь идет о диагностике хромосомных аномалий, морфологические нарушения эмбриогенеза мы можем увидеть только на УЗИ)
К сожалению, пока данные анализы приходится проводить в лабораториях США, поэтому стоимость их достаточно высока, но в скором времени они будут выполняться в России и, надеюсь, станут более доступны для наших сограждан.
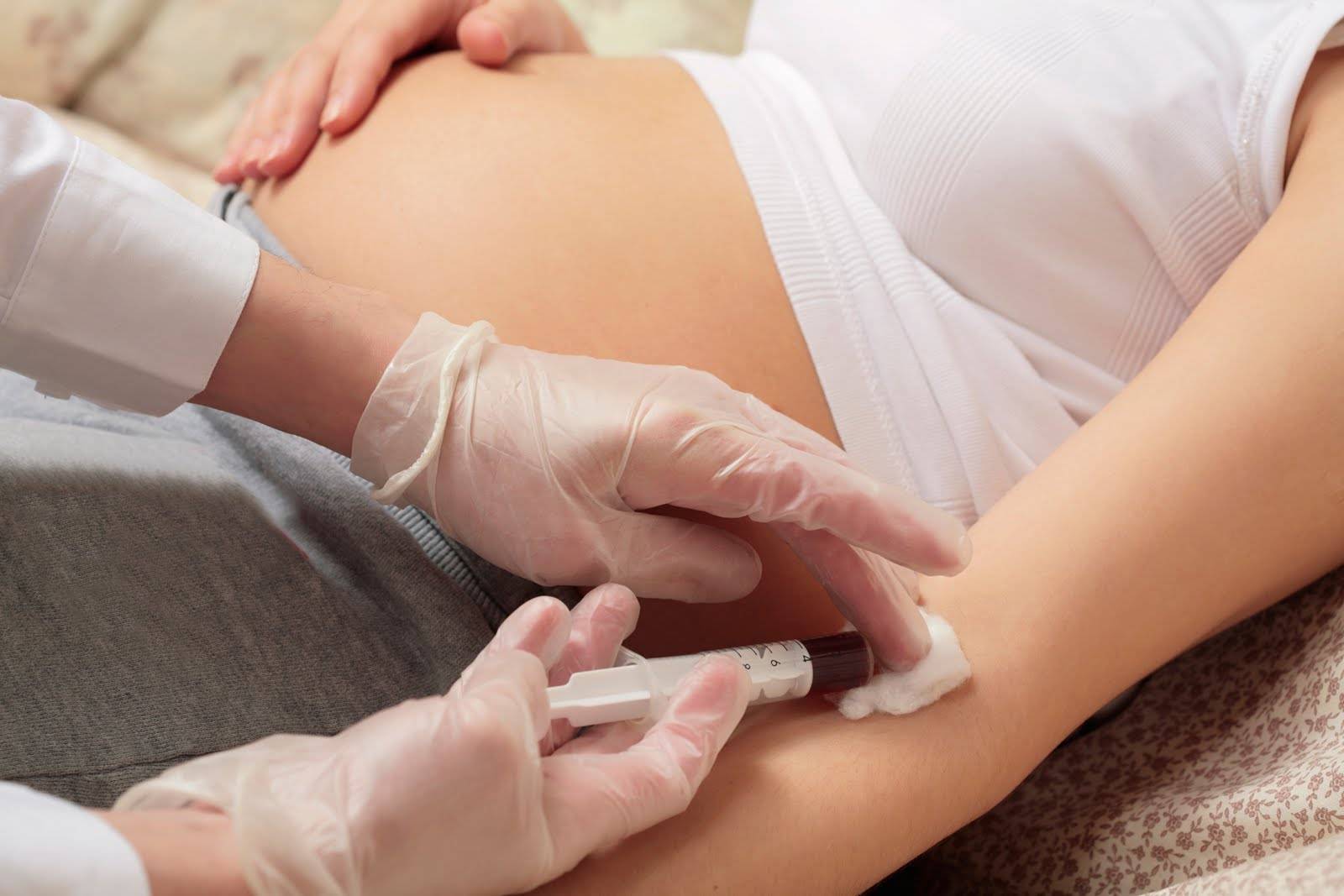
Диагностика
О генетических патологиях чаще всего можно узнать, пока женщина еще вынашивает ребенка. Это касается и трисомий. Скрининг беременности проводят с 11-й по 13-ю неделю. Женщина сдает анализы крови (биохимию), проводится УЗИ. Также диагностика заключается в определении каротипа эмбриона, если женщина находится в группе риска (отягощенный семейный анамнез, инфекционные болезни в первом триместре и т. д.).
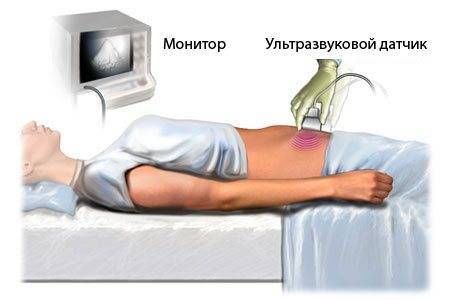
В скрининге первого семестра определяют, сколько в крови хорионического гормона человека и плазменного протеина А ассоциированного с беременностью. Потом учитывают возраст беременной, чтобы узнать, с каким риском у нее может родиться ребенок с трисомией 18.








Если женщину отнесли в группу риска, чуть позже делают биопсию плода, чтобы точно знать, родится ли ребенок с отклонениями, или здоровый. С 8 до 12 неделю берется анализ ворсин хориона. С 14 по 18-ю неделю проводится изучение вод, окружающих плод. После 20-й недели могут сделать кордоцентез. Процедура подразумевает, что возьмут крови из пуповины (в процессе применяется ультразвук для контроля взятия материала).
В материале обнаруживают количество хромосом. В этом помогает метод КФ–ПЦР. При условии непрохождения беременной генетического скрининга на поздник сроках гестации делают предварительную диагностику генетической мутации методом ультразвукового исследования. Во втором и третьем триместре есть признаки, которые говорят о том, что ребенок с большой вероятностью родится с трисомией:
- заячья губа
- низко расположенные уши плода
- микроцефалия
- волчья пасть
- пороки опорно–двигательного аппарата
- пороки развития мочеполовой системы
- пороки сердца и сосудов
Диагностические признаки синдрома у ребенка
После рождения ребенка опорными диагностическими признаками наличия синдрома Эдвардса являются следующие:
- Микроцефалия, малый вес при рождении
- Наличие «заячьей губы» или «волчьей пасти»
Признаки характерной дерматографической картины:
- неразвитая на пальцах дистальная сгибательная складка
- наличие в 1/3 случаев поперечной ладонной борозды
- дуги на подушечках пальцев рук
- изменение кожного рисунка ладони: дистальное расположение осевого трирадиуса и увеличение гребневого счета.
Далее диагноз также подтверждается с помощью определения кариотипа ребенка методом КФ–ПЦР.
Синдром Эдвардса на УЗИ — кисты сосудистых сплетений
На ранних сроках синдром Эдвардса на УЗИ заподозрить крайне трудно, однако в 12 недель беременности уже выявляют характерные для него симптомы косвенного характера
- Признаки задержки развития плода
- Брадикардия (снижение у плода частоты сердечных сокращений)
- Омфалоцеле (наличие грыжи брюшной полости)
- Отсутствие визуализации косточек носа
- В пуповине одна, а не 2 артерии
Также на УЗИ могут быть обнаружены кисты сосудистых сплетений, представляющих собой полости с содержащейся в них жидкостью. Сами по себе они не несут угрозу для здоровья и исчезают к 26–недельному сроку беременности. Однако такие кисты очень часто сопровождают различные генетические заболевания, например, синдром Эдвардса (в данном случае кисты обнаруживаются у 1/3 детей, страдающих этой патологией), поэтому при обнаружении таких кист врач направит беременную женщину на обследование в генетическую консультацию.
Хромосомная патология – синдром Шерешевского-Тернера
Риск хромосомной патологии плода – синдрома Шерешевского-Тернера составляет 1 к 3500. Кариотип заболевания – 45.Х. Характеризуется патология антимонголоидным разрезом глаз, в 65% случаев встречается лимфатический отек стоп, голеней, кистей рук у новорожденного младенца, которые могут проявляться в течении первых месяцев жизни малыша. Патология имеет выраженные признаки – короткая шея, которая встречается в половине случаев патологии, крыловидные складки (шея сфинкса) от затылка до надплечья, встречаются в 65% случаев заболевания. У всех детей с синдромом Шерешевского-Тернера маленький рост, бочкообразная грудь с широко расставленными сосками встречается в 55% случаев. При кариотипе 45.Х у всех больных детей диагностируется половой инфантилизм. Патология характеризуется недоразвитием молочных желез, аменореей, эмоциональной бедностью. Патологию лечат стимуляцией роста ребенка, формированием менструального цикла с помощью гормональной терапии, по показаниям применяют хирургическое лечение, психотерапевтическое лечение.
ЗАКЛЮЧЕНИЕ
Представленные нами клинические случаи демонстрируют фенотипическое разнообразие синдрома 18р- и 18q-. Общими для всех наших пациентов признаками явились наличие дефицита гормона роста, дисморфия лица и задержка речевого развития.
Врожденный гипопитуитаризм представляет собой большую опасность, особенно в неонатальном периоде, так как дефицит кортизола, равно как и дефицит гормона роста, могут приводить к развитию тяжелых рецидивирующих гипогликемий у детей в первые дни и месяцы жизни. Наличие гипогликемического синдрома в период новорожденности в сочетании с холестазом является абсолютным показанием для проведения исследования тропных гормонов гипофиза, позволяющего в максимально ранние сроки верифицировать диагноз и своевременно начать заместительную гормональную терапию.
Врожденный гипопитуитаризм может иметь различную этиологию. Наличие стигм дисэмбриогенеза у детей с полным или частичным выпадением тропных гормонов гипофиза должно служить поводом к проведению кариотипирования или хромосомного микроматричного анализа для исключения патологии 18 хромосомы.





