

Причины снижения гематокрита
Гестационный период. Начиная со второго триместра, происходит увеличение объема циркулирующей крови, а число красных кровяных клеток не изменяется.
Гипергидратация. Внутривенные массивные инфузии приводят к увеличению объема циркулирующей крови, то есть к разведению крови, тогда как количество эритроцитов остается прежним.
Анемии различного генеза. Недостаток эритроцитов закономерно сказывается на показателях гематокрита.
Интенсивное разрушение эритроцитов. Происходит при наличии гемолитических анемий наследственного либо приобретенного генеза, при отравлении гемолитическими токсинами (соли тяжелых металлов, потребление бледной поганки) или серьезных инфекционных болезнях (малярия, брюшной тиф).
Кровопотеря (внутреннее либо наружное кровотечение). Восполнение объема циркулирующей крови можно произвести быстро кристаллическими или коллоидными растворами. Восстановление утраченных эритроцитов происходит лишь через определенное время либо путем гемотрансфузии.
Гиперпротеинемия (повышение общего белка в крови). Наблюдается данное состояние при миеломе, лимфогранулематозе, парапротиенемичских гематобластозах, поносах и рвоте. Избыток белка в крови оттягивает жидкости из организма в кровяное русло, увеличивая объем циркулирующей крови с сохранением прежнего числа эритроцитов.
Расстройство образования новых эритроцитов в костном мозге. Может быть обусловлено болезнями крови (анемии, лейкемии), приемом цитостатиков и противоопухолевых средств, парезом почек.
Тактика в зависимости от степени снижения показателя:
- Снижение гематокрита в диапазоне 35-30% требует амбулаторного наблюдения врача и изменения рациона питания (увеличить потребление мяса, листовой зелени, печени).
- При уменьшении гематокритной величины до 29-24% назначается прием железосодержащих средств, витаминов группы В и фолиевой кислоты.
- Если показатель снижается до 13% и менее, состояние расценивается как крайне тяжелое и требует экстренной госпитализации.
При снижении гематокрита и выявлении анемии у людей старше 65 лет, обследование должно быть более тщательным, поскольку анемия в пожилом возрасте часто сопутствует серьезным заболеваниям, например, почечной недостаточности, гипо- или гипертиреозу.
Клиническая картина
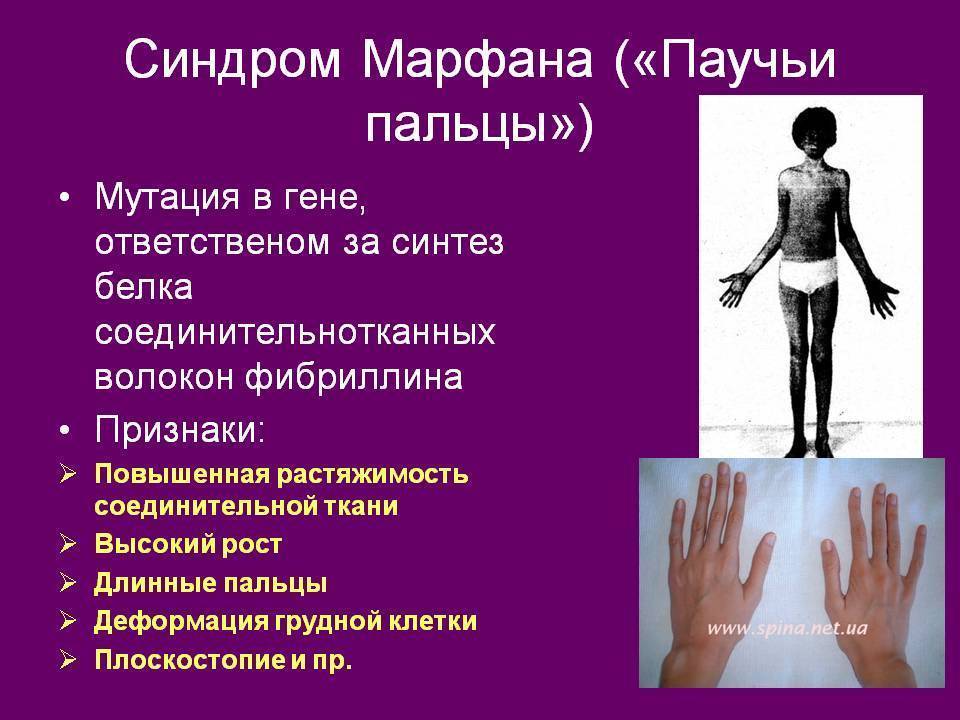
Для данного заболевания характерен внешний вид больного. Мальчики при рождении имеют длину тела около 53 см, а во взрослом возрасте достигают 190 см. Девочки рождаются с длиной тела 52,5 и вырастают до 175 см. Таким образом, больные данным генетическим заболеванием имеют высокий рост. Помимо роста, у детей отмечают непропорционально длинные конечности и короткое тело, удлинённые паукообразные пальцы, вытянутое лицо и форма черепа, астеническое телосложение, снижение подкожно-жировой клетчатки.
Для синдрома Марфана характерно многообразие клинических симптомов.
Поражение костной системы
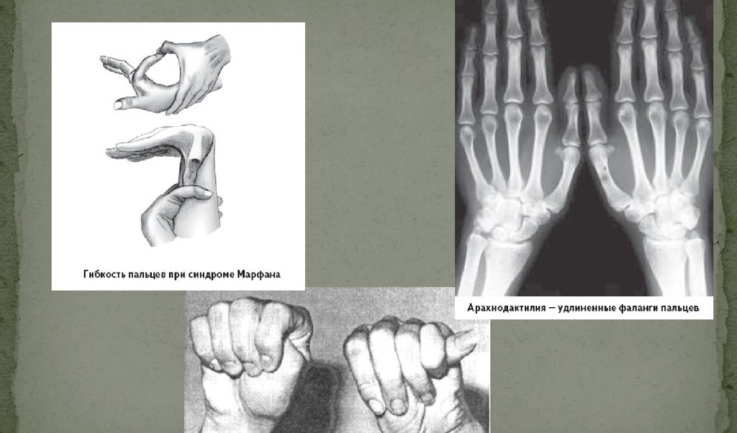
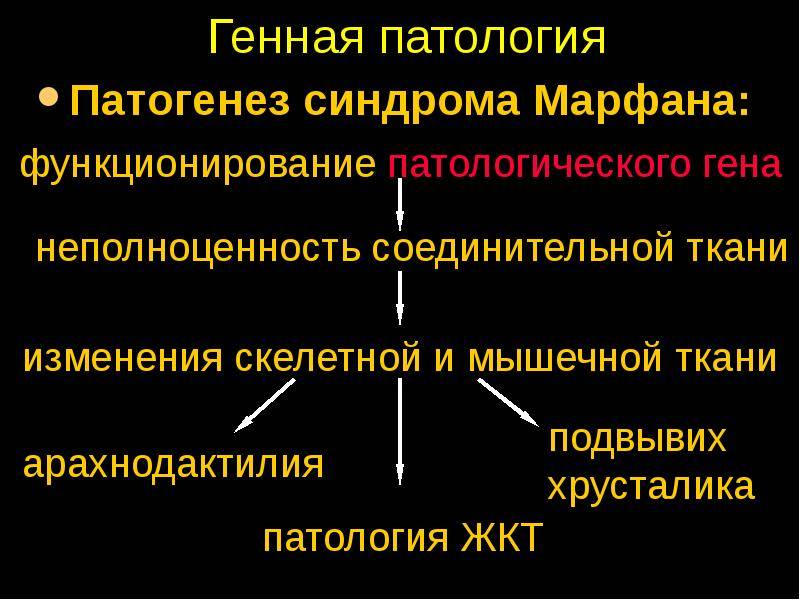
Наиболее часто поражается при заболевании именно костная система. В симптомокомплекс можно включить высокий рост, непропорциональное тело, арахнодактилия (паукообразные пальцы). У детей с данным синдромом развивается сколиоз (искривление позвоночника в сторону). Нередко выявляется деформация грудной клетки, изменения могут быть как внутрь (воронкообразная грудь), так и наружу (килевидная). Высокое нёбо, неправильный прикус у детей развивается, так как форма черепа имеет вытянутую форму. Такие изменения лицевого скелета способствуют нарушению речи. Плоскостопие также является симптомом болезни Марфана. У детей отмечается гипермобильность суставов (чрезмерная гибкость). Пациенты могут предъявлять жалобы на боль в суставах и костях.
Поражение глазного аппарата
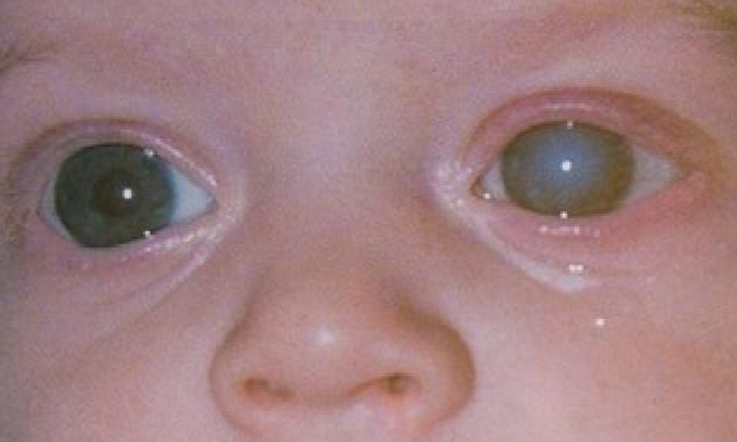
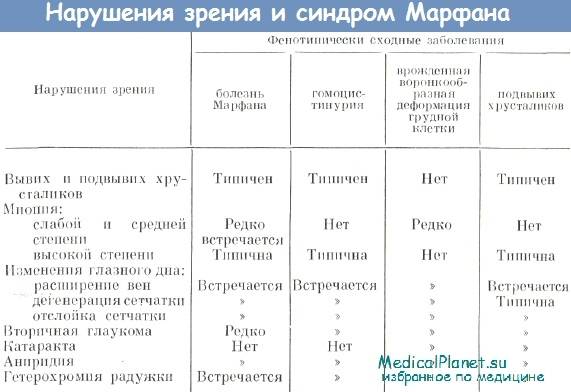
У 80% больных синдромом Марфана выявляют изменение положения хрусталика.
Наиболее часто у детей офтальмолог при помощи щелевой дампы выявляет подвывих хрусталика. Помимо дислокации хрусталика, выявляется астигматизм, близорукость, глаукома в раннем возрасте.
Поражение сердечно-сосудистой системы
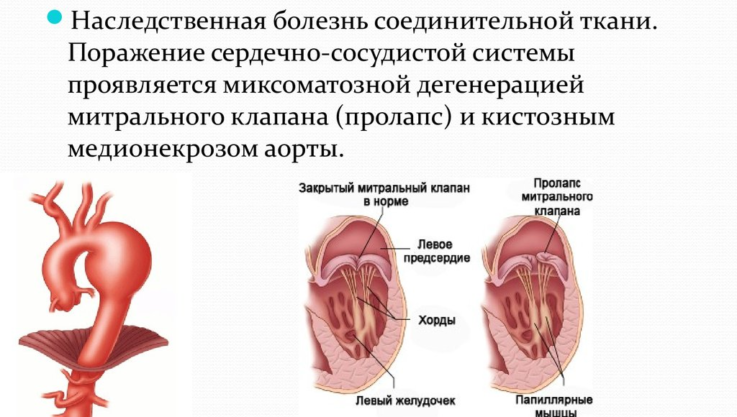
Самыми грозными являются изменения со стороны сердечно-сосудистой системы. Дети могут жаловаться на повышенную утомляемость, учащение сердцебиения и дыхания, нарушение ритма сердца, возможно появление болей в области сердца. Появление шумов при аускультации даёт повод для более тщательного обследование пациента. Наиболее опасным является изменение аорты: аневризмы (выпячивание стенки) или расслоение верхней её части. Но такие явления никак себя не проявляют. Особого внимания требуют беременные женщины, так как во время родов возможно расслоение аорты и смертельный исход. Поэтому необходимо каждые 6 недель проводить ультразвуковое исследование сердца и сосудов, для динамических замеров диаметра корня аорты.
Поражение лёгких
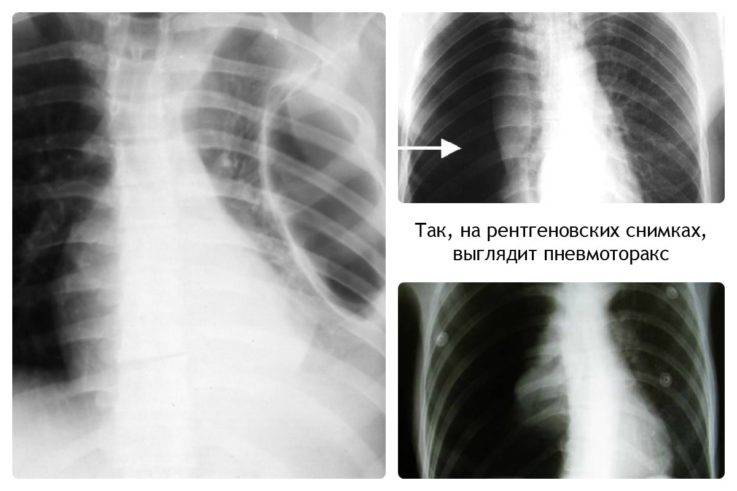
При синдроме Марфана возникает риск возникновение спонтанного пневмоторакса. Такое состояние характеризуется изменением давления в плевральной полости. Пациент чувствует боль в грудной клетке, наблюдается одышка, побледнение кожного покрова. Ребёнок не может глубоко вдохнуть. При отсутствии лечения пневмоторакс приводит к смерти больного.
Поражение нервной системы
При синдроме Марфана поражается твёрдая оболочка спинного мозга, а именно происходит её ослабление или растяжение (эктазия). Такое изменение несёт в себе целый ряд клинических проявлений: головная боль, боль в спине и конечностях. На рентгенологическом исследовании такие изменения на ранних стадиях не видны, поэтому диагностический поиск может зайти в тупик. Данное поражение может наблюдаться лишь при более эффективных методиках обследования пациента, таких как магнитно-резонансная и компьютерная томография.
Лечение Болезни (синдрома) Марфана:
К терапии синдрома Марфана относится консервативная и хирургическая коррекция сердечно-сосудистых нарушений, поражений скелета и органы зрения.
Лечение, прежде всего, направлено на профилактику развития заболевания и осложнений в сердечно-сосудистой системе. Если диаметр аорты до 4 см больному назначают антагонисты кальция, β-адреноблокаторы или ингибиторы АПФ. Хирургическое вмешательство проводится только, когда диаметр аорты составляет больше 5 см при пролапсе митрального клапана, недостаточности клапанов сердца, восходящей части и расслоении аорты.
При синдроме Марфана выполняют реконструктивные операции на аорте. В случае необходимости выполняется протезирование митрального клапана.
Больным с синдромом Марфана назначается коррекция зрения методом подбора очков и контактных линз, а в сложных случаях – путем лазерного или хирургического лечения болезней зрения.
Детям со скелетными нарушениями проводят хирургическую стабилизацию позвоночника, эндопротезирование тазобедренных суставов, торакопластику.
В курс лечения входит: витаминотерапия, патогенетическая коллагеннормализующая и метаболическая терапия.
Болезни радужной оболочки
Синдрому Марфана нередко сопутствуют изменения в радужной оболочке — это связано с повышенной растяжимостью тканей. Могут возникнуть колобомы, гипоплазия или атрофия радужки с нарушением ее диафрагмальной функции. Колобома — это дефект, проявляющийся в отсутствии части глазной оболочки. Обычно имеет грушевидную форму и располагается в нижней части радужки. К наиболее ранним проявлениям у людей с синдромом Марфана относят гипоплазию стромы радужки, особенно ее пигментной зрачковой каймы. Слабость дилататора (мышцы-расширителя) у больных не позволяет достичь полного расширения зрачка даже с помощью мидриатиков.
Прогноз
В связи с возможностью осложнений, включая и почечную недостаточность, которые могут наблюдаться и без операции, прогноз при Марфана синдроме во всех случаях, как правило, неблагоприятен.
Библиография: Семячкина А. Н. и Барашнев Ю. И. Принципы диагностики болезни Марфана, Педиатрия, № 3, с. 58, 1974, библиогр.; Фадеева М. А. и др. Синдром Марфана у детей, в кн.: Наследственная и приобретенная патол. обмена веществ у детей, под ред. В. А. Таболина, с. 122, М., 1971; Вrentоn D. Р. а. о. Homocystinuria and Marfan’s syndrome. J. Bone Jt Surg., v. 54-B, p. 277, 1972; Maifan B. J. Un cas de deformation congenitale des quatre membres, plus prononcee aux extremites, characterisee par l’allongement des os avec un certain degre d’amincissement, Bull. Soc. med. Hop. Paris, t. 13, p. 220, 1896.
H. И. Кондрашин.
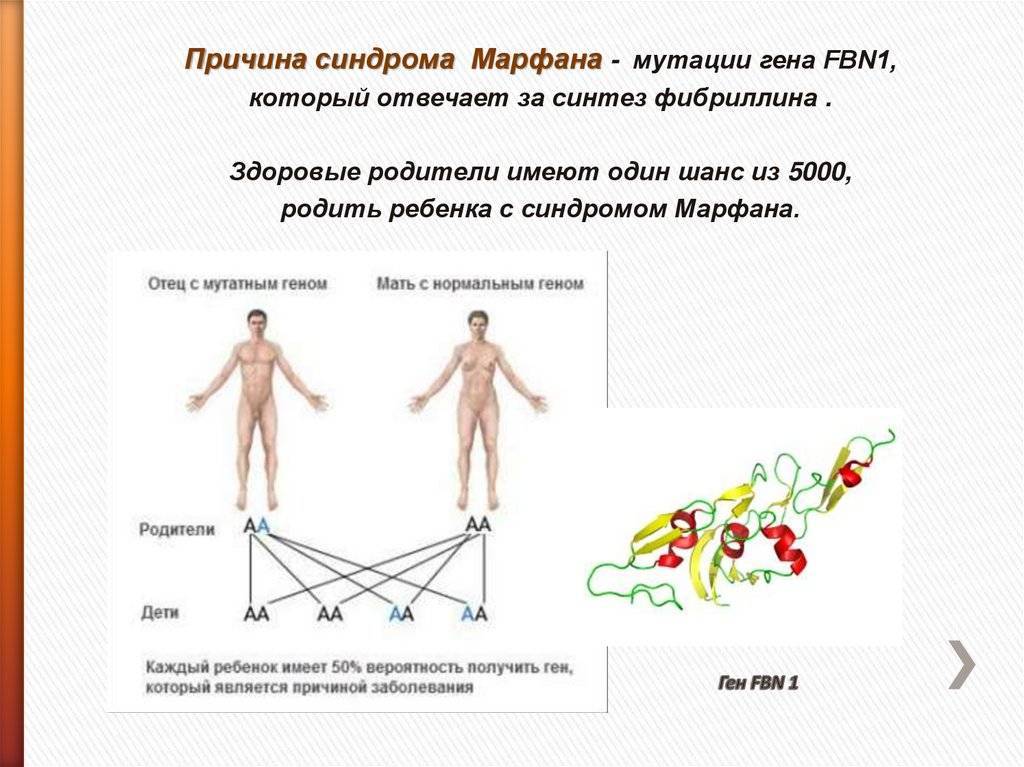
Причины синдрома Марфана
Данное генетическое заболевание вызвано дефектом гена FBN1 в длинном плече 15 хромосомы. Этот ген кодирует белок гликопротеин фибриллин-1, который отвечает за прочность и эластичность соединительной ткани. Соответственно, все проявления патологии связаны с тем, что соединительнотканные структуры в организме человека теряют свои нормальные свойства.
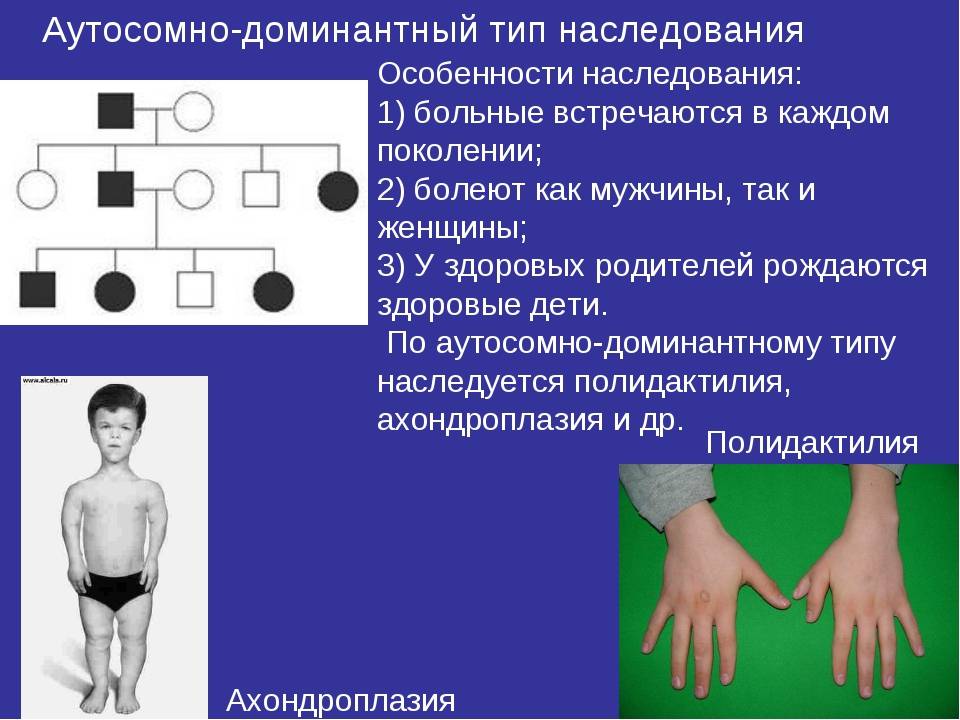
Наследуется мутация по аутосомно-доминантному признаку, то есть дети получают патологический ген от родителей, которые страдают от патологии. При этом шанс ребенка получить мутацию от одного из родителей составляет 50% (рис. 1). Синдром не передается через поколение: здоровые дети больных родителей не могут передать ген своим потомкам.
Однако примерно у 25% людей с синдромом Марфана никто из родителей не оказывается носителем аномалии гена FBN1: в таком случае мутация развивается спонтанно.
До сих пор не выявлено определенных факторов риска развития этого генетического нарушения: заболевание встречается одинаково часто среди мужчин и женщин, а его распространенность не зависит от расы или этнической группы. Частота заболеваемости у этой патологии составляет примерно 1 случай на .







Если клинические признаки мутации ярко выражены, заподозрить болезнь можно уже в первые месяцы жизни ребенка, но стертые формы заболевания часто проявляются уже во взрослом возрасте, когда пациент обращается к врачам по поводу различных проявлений синдрома.
Важно! Не стоит записываться на генетическое обследование в качестве медосмотра. Поиски «поломки» гена FBN1 оправданы только в случае, если болезнь проявляет себя характерными признаками: бессимптомное носительство этой мутации невозможно
Если у одного из родителей установлен этот диагноз, будущей маме следует пройти генетическое обследование еще до родов. Это позволит заранее узнать, передалась ли аномалия ребенку.
Диагностика
УЗИ человека с синдромом Марфана, показывающее расширенный корень аорты
Диагностические критерии MFS были согласованы на международном уровне в 1996 году. Однако синдром Марфана часто трудно диагностировать у детей, поскольку они обычно не проявляют симптомов до достижения полового созревания. Диагноз основывается на семейном анамнезе и комбинации основных и второстепенных показателей расстройства, редко встречающихся в общей популяции, которые встречаются у одного человека, например: четыре скелетных признака с одним или несколькими признаками в другой системе организма, такой как глазная и сердечно-сосудистые у одного человека. Следующие состояния могут быть результатом MFS, но могут также возникать у людей без каких-либо известных основных заболеваний.
- Аневризма или расширение аорты
- Арахнодактилия
- ГЭРБ
- Двустворчатый аортальный клапан
- Кисты
- Кистозный медиальный некроз
- Дегенеративная болезнь диска
- Искривленная перегородка
- Дуральная эктазия
- Ранняя катаракта
- Ранняя глаукома
- Ранний остеоартроз
- Эктопия лентис
- Эмфизема
- Ирис колобома
- Выше среднего роста
- Учащенное сердцебиение
- Грыжи
- Небо с высокой аркой
- Гипермобильность суставов
- Кифоз (сгорбившись)
- Негерметичный сердечный клапан
- Неправильный прикус
- Микрогнатия (малая нижняя челюсть)
- Пролапс митрального клапана
- Миопия (близорукость)
- Обструктивная болезнь легких
- Остеопения (низкая плотность костной ткани)
- Pectus carinatum или экскаватум
- Pes planus ( плоскостопие )
- Пневмоторакс (коллапс легкого)
- Отслойка сетчатки
- Сколиоз
- Апноэ во сне
- Растяжки не от беременности или ожирения
- Зубы скучены
- «Узкое, худое лицо»
- Дисфункция височно-нижнечелюстного сустава (ВНЧС)
Пересмотренная гентская нозология
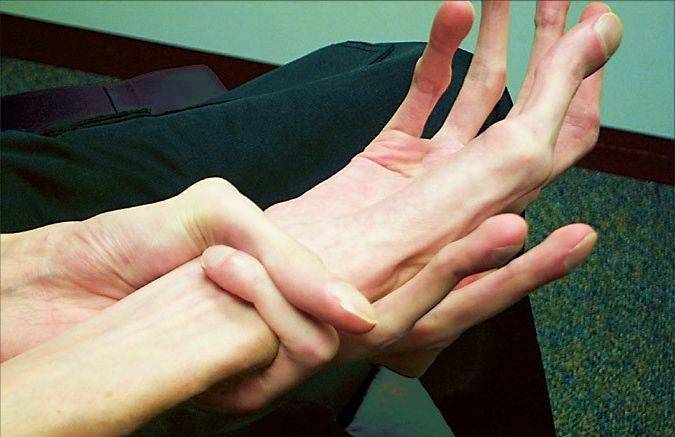
Знак большого пальца; верхний : нормальный, нижний : синдром Марфана
В 2010 году нозология Гента была пересмотрена, и новые диагностические критерии заменили предыдущее соглашение, заключенное в 1996 году. Семь новых критериев могут привести к постановке диагноза:
При отсутствии семейного анамнеза MFS:
- Z-показатель корня аорты ≥ 2 И эктопия lentis
- Z-показатель корня аорты ≥ 2 И мутация FBN1
- Z-оценка корня аорты ≥ 2 И системная оценка *> 7 баллов
- Ectopia lentis И мутация FBN1 с известной патологией аорты
При наличии семейного анамнеза MFS (как определено выше):
- Эктопия лентис
- Системный балл * ≥ 7
- Z-показатель корня аорты ≥ 2
- Очки за системный балл:
- Знак запястья И большого пальца = 3 (знак запястья ИЛИ большого пальца = 1)
- Деформация Pectus carinatum = 2 (асимметрия грудной клетки или грудной клетки = 1)
- Деформация заднего отдела стопы = 2 (плоская стопа = 1)
- Эктазия твердой мозговой оболочки = 2
- Протрузия вертлужной впадины = 2
- пневмоторакс = 2
- Уменьшение соотношения верхнего и нижнего сегментов И увеличение руки / роста И отсутствие тяжелого сколиоза = 1
- Сколиоз или грудопоясничный кифоз = 1
- Уменьшенное разгибание локтей = 1
- Черты лица (3/5) = 1 ( долихоцефалия , энофтальм , опускание глазных щелей , гипоплазия скуловой кости , ретрогнатия )
- Бороздки кожи ( растяжки ) = 1
- Близорукость > 3 диоптрии = 1
- Пролапс митрального клапана = 1
Знак большого пальца (знак Стейнберга) вызывается просьбой человека согнуть большой палец как можно дальше, а затем сомкнуть пальцы над ним. Положительный признак большого пальца – это когда вся дистальная фаланга видна за локтевым краем кисти, что вызвано сочетанием гипермобильности большого пальца, а также большого пальца, который длиннее обычного.
Знак запястья (знак Уокера-Мердока) вызывается, когда человека просят обхватить большим пальцем и пальцами одной руки другое запястье. Положительным признаком запястья является перекрытие мизинца и большого пальца из-за сочетания тонких запястий и длинных пальцев.
Дифференциальная диагностика
Многие другие расстройства могут вызывать те же характеристики тела, что и синдром Марфана. Генетическое тестирование и оценка других признаков и симптомов могут помочь их дифференцировать. Ниже приведены некоторые расстройства, которые могут проявляться как «марфаноид»:
- Врожденная контрактурная арахнодактилия , также известная как синдром Билса-Хехта
- Синдром Элерса-Данлоса
- Гомоцистинурия
- Синдром Лойса-Дитца
- MASS фенотип
- Множественная эндокринная неоплазия 2В типа
- Синдром Шпринцена – Гольдберга
- Синдром Стиклера
Примечания
- Marfan A. B. Un cas de deformation congenital des quatre membres plus prononcee aux extremities caracterisee par l’allongement des os avec un certain degre d’amincissement. // Bulletins Et Memoires De La Societe Medicale Des Hopitaux De Paris. — 1896. — Vol. 13. — PP. 220—226.
- Pyeritz R.E. Disorders of fibrillins and microfibrilogenesis: Marfan syndrome, MASS phenotype, contratural arachnoductyly and related conditions. In: Rimoin D.L., Connor J.M., Pyeritz R.E. (eds). «Principles and Practice of Medical Genetics», 3rd ed. New York: Churchill Livingstone, in press 1996.
- Goldman’s Cecil Medicine 978-1-4557-1167-3,
Советы родителям ребенка с синдромом Марфана
Конечно, грустно и горько осознавать, что у ребенка имеется такое опасное заболевание. Однако только правильное поведение родителей поможет преодолеть трудности, в том числе создать нужный психологический настрой. Нужно научиться жить с этой проблемой и постоянно держать под контролем состояние ребенка
Важно найти хороших специалистов, которые будут заниматься его здоровьем
Следует также научить его относиться с достоинством к своему состоянию, не реагировать на возможные насмешки со стороны. Лучше привить ему любовь к занятиям, которыми он сможет заниматься в дальнейшем: пусть это будет программирование или, например, музыка. Если диагноз был поставлен уже в старшем возрасте, то придется отказаться от некоторых видов спорта.

Важно также наладить хороший контакт с учителями, объяснив им, что это заболевание предполагает некоторые особенности. Например, сидеть ему лучше за первой партой из-за проблем со зрением, не стоит испытывать слишком сильных физических нагрузок
Каждому родителю хочется обеспечить своему ребенку счастливое детство. Дети с синдромом Марфана должны знать, что в мире много интересных занятий, которые им доступны.
Лечение синдрома Марфана
Специфической терапии, направленной на устранение причины заболевания не существует. В настоящее время не разработаны методы влияния на наследственный аппарат клетки. Поэтому основная цель лечения синдрома Марфана – предотвращение прогрессирования заболевания, борьба с симптомами болезни:
патология сердца и сосудов.
Самым опасным проявлением недуга считается аневризма, расширение участка аорты. Коварство болезни заключается в непрерывном, длительном прогрессировании симптомов
Случается, что опасный симптом формируется к 18 годам, поэтому важно уделять достаточно внимания ежегодному обследованию и лечению сердечно-сосудистой системы
Грубые пороки развития сердца и сосудов, тяжёлые осложнения хронических болезней лечатся оперативно. Из лекарственных препаратов назначаются ингибиторы АПФ, блокаторы кальциевых каналов. Применение b-адреноблокаторов (пропанолола, атенолола) показано при расширении корня аорты, пролапсе клапанов, аритмиях.
Назначение препаратов и подбор необходимой дозировки должно проводиться врачом-кардиологом с учётом данных обследования ребёнка. Необоснованное назначение лекарственных средств может привести к ухудшению состояния ребёнка;

болезни опорно-двигательного аппарата.
Существую исследовании, указывающие на дефицит некоторых макроэлементов (кальций, цинк, кобальт, магний) и белков, необходимых для строительства соединительной ткани при синдроме Марфана. Поэтому для предотвращения прогрессирования патологии назначаются витаминно-минеральные комплексы, гиалуроновая кислота, викасол, колекальциферол. Оперативное лечение показано при грубых патологиях развития скелета;
заболевания органов зрения.
Исправление патологии зрения проводится с помощью подбора специальных очков, контактных линз, оперативного лечения катаракты и глаукомы, смещения хрусталика.
Опасным осложнением синдрома является отслойка сетчатки. Эта патологии возникает при активном занятии спортом у ребят с дефектом соединительной ткани. Повышенная физическая нагрузка, прыжки, травмы приводят к отделению тонкой сетчатой оболочки от сосудистой. Такое нарушение сопровождается резким снижением остроты зрения, которое не всегда является обратимым. Поэтому ребятам с генетическим синдромом следует избегать чересчур активных занятий, для таких детей хорошо подходит плавание в бассейне;
нарушенный обмен веществ.
Для улучшения метаболизма рекомендовано использовать в комплексном лечении аскорбиновую и янтарную кислоты, карнитин, препараты магния, токоферола ацетата. С целью нормализации обмена хрящевой ткани используются глюкозаминсульфат, хондроитинсульфат.
Существуют данные, указывающие на необходимость введения диеты с высоким содержанием магния для детей с синдромом Марфана. Этот элемент помогает справиться с повышенным содержанием катехоламинов в крови и способствует восстановлению дефектов кровеносных сосудов. Хорошо включать в ежедневный рацион орехи, какао, гречневую и ячневую каши, сухофрукты.
Что происходит при синдроме Марфана?
У всех есть белок фибриллин-1, который образует эластичные волокна в соединительной ткани. Фибриллин-1 также влияет на другой белок в вашем организме, трансформируя бета-фактор роста (TGF-бета), который помогает контролировать рост и развитие организма. Люди с синдромом Марфана наследуют мутацию гена, которая изменяет то, как организм использует фибриллин-1, что приводит к избытку факторов роста, это вызывает:
- Ткани кровеносных сосудов, сердца, связок, сухожилий и хрящей растягиваются сильнее, что делает их слабыми.
- Необычно сильный рост костей, делая их длиннее, чем обычно.
Синдром Марфана у детей
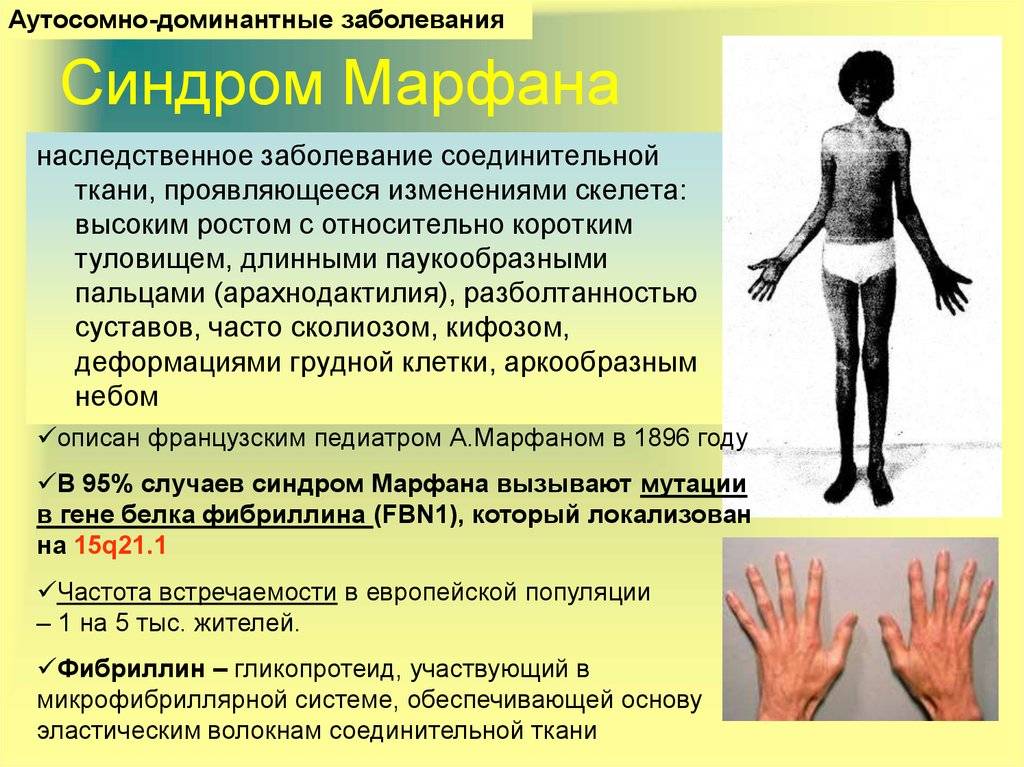
Синдром Марфана (врожденная мезодермальная дистрофия) — это аутосомно-доминантное генетическое заболевание соединительной ткани с преимущественным поражением скелета (удлинение трубчатых костей, долихостеномелия, арахнодактилия, гипермобильность суставов), глаз (миопия, подвывих хрусталика) и сердечно-сосудистой системы (пролапс митрального клапана, расслаивающая аневризма аорты). Впервые данная патология была описана в 1896 году французским педиатром А. Марфаном, который выявил характерную деформацию скелета у 5-летней девочки (длинные трубчатые кости, паукообразные пальцы рук, высокорослость). Болезнь Марфана возникает в результате нарушения синтеза фибриллина-1, который является основным структурным белком соединительной ткани. Данный синдром обусловлен мутацией гена, кодирующего продукцию этого гликопротеина.










Синдром Марфана у детей относится к редким врожденным аномалиям. Частота диагностированных случаев составляет 1:10 000 – 1:20 000. От болезни Марфана чаще страдают мальчики.
Для данной патологии характерны следующие признаки:
- аутосомно-доминантный тип наследования (если один из родителей имеет данное заболевание, то риск возникновения его у ребенка колeблется от 50 до 100%);
- высокая пенетрантность (болезнь фенотипически проявляется при небольшом количестве копий аллелей мутантного гена);
- различная экспрессивность (трaнcформация гена в определенную белковую структуру).
Кроме семейного наследования, синдрома Марфана в 25% случаев возможна первичная мутация гена, что объясняет рождение больного ребенка у здоровых родителей.
Среди известных мировых личностей синдромом Марфана болели Авраам Линкольн, Ганс Христиан Андерсен, Сергeй Рахманинов, а американскому пловцу Майклу Фелпсу данный генетический порок дал большое преимущество перед другими спортсменами, что позволило ему стать 23-кратным олимпийским чемпионом. Некоторые ученые утверждают, что люди с болезнью Марфана владеют уникальным интеллектом. Данный факт стал основанием для поиска корреляционной связи между геном данного заболевания и геном «гениальности».
Почему при определении признаков синдрома Марфана нужно обратиться к врачу?
Сама по себе генетическая аномалия совместима с жизнью. Однако опасны последствия болезни, вызванной FBN1 мутацией:
- разрывы крупных сосудов, чаще всего — аорты;
- хроническая сердечная недостаточность — неспособность сердца обеспечивать необходимую работу для кровоснабжения всех органов;
- снижение остроты зрения или полная потеря зрительной функции.
Разрыв аневризмы аорты или другого магистрального сосуда часто заканчивается моментальным летальным исходом. Хроническая сердечная недостаточность может перейти в острую форму, а без экстренной медицинской помощи также привести к фатальным последствиям — внезапной коронарной смерти. Именно эти осложнения чаще всего приводит к гибели детей с синдромом Марфана. Особая опасность ждет женщину с синдромом мутации гена FBN1 во время беременности: повышенная нагрузка на аорту в разы увеличивает риск ее разрыва.
Чтобы предупредить развитие опасных осложнений и компенсировать возникающие нарушения, родителям нужно как можно раньше обратиться за медицинской помощью при первом подозрении на синдром Марфана у ребенка
При этом важно не только однократно провести обследование, но и стать на учет к врачам, которые занимаются коррекцией проявлений синдрома:
- специалисту по генетическим болезням;
- кардиологу;
- ортопеду-вертебрологу;
- дерматологу;
- офтальмологу;
- гастроэнтерологу.
Список специалистов зависит от степени выраженности заболевания, при этом регулярно необходимо проходить комплексные профилактические осмотры для раннего выявления новых нарушений.
Синдром Марфана — болезнь гениев?
С синдромом Марфана связаны не только многочисленные поводы для обращения к врачам. Часто люди с мутацией гена FBN1 компенсируют физические проявления болезни интеллектуальными способностями, поэтому это генетическое заболевание даже называют «синдромом гениев». Считается, что повышенный выброс адреналина из-за патологических изменений в надпочечниках определяет высокий тонус умственной и психической активности у таких пациентов. Именно поэтому в числе людей с синдромом Марфана можно найти известных личностей. Например, Юлию Цезарю, Аврааму Линкольну и Шарлю де Голлю патология не помешала стать известными политическими деятелями; Ганс Христиан Андерсен и Корней Чуковский создали уникальные литературные произведения, а Никколо Паганини прославился как гениальный музыкант.
Современные знаменитости также не скрывают свои недостатки и становятся еще более популярными из-за генетического дефекта. Например, солисту американской рок-группы Deerhunter Брэдфорду Коксу нетипичная внешность придает особый шарм, а испанский актер Хавьер Ботет очень востребован, поскольку правдоподобно и талантливо играет отрицательных героев в голливудских фильмах ужасов (рис. 6).
Советы родителям ребенка с синдромом Марфана
Конечно, грустно и горько осознавать, что у ребенка имеется такое опасное заболевание. Однако только правильное поведение родителей поможет преодолеть трудности, в том числе создать нужный психологический настрой. Нужно научиться жить с этой проблемой и постоянно держать под контролем состояние ребенка
Важно найти хороших специалистов, которые будут заниматься его здоровьем
Следует также научить его относиться с достоинством к своему состоянию, не реагировать на возможные насмешки со стороны. Лучше привить ему любовь к занятиям, которыми он сможет заниматься в дальнейшем: пусть это будет программирование или, например, музыка. Если диагноз был поставлен уже в старшем возрасте, то придется отказаться от некоторых видов спорта.

Важно также наладить хороший контакт с учителями, объяснив им, что это заболевание предполагает некоторые особенности. Например, сидеть ему лучше за первой партой из-за проблем со зрением, не стоит испытывать слишком сильных физических нагрузок. Каждому родителю хочется обеспечить своему ребенку счастливое детство
Дети с синдромом Марфана должны знать, что в мире много интересных занятий, которые им доступны
Каждому родителю хочется обеспечить своему ребенку счастливое детство. Дети с синдромом Марфана должны знать, что в мире много интересных занятий, которые им доступны.
Причины
Основная причина, которая вызывает развитие данного синдрома является мутации в генах, которые кодируют синтез фибриллина-1, гликопротеина и является плейотропным. Фибриллин является белком межклеточного матрикса, который придает сократимость и эластичность соединительной ткани. В результате мутации происходит нарушение формирования волокнистых структур, что приводит к потери упругости и прочности соединительной ткани.
Причины, которые могут способствовать развитию этого синдрома:
- наследственность (наследуется по аутосомно-доминантному типу);
- различные мутации (более 1000 видов) в гене FBN1 (отвечает за синтез фибриллина);
- различные мутации гена, который трансформирует фактор роста – TGFBR-2;
- генетическая предрасположенность;
- возраст отца (возможно появление данного синдрома у ребенка, если его отцу больше 35 лет).
Диагностика
При подозрении на развитие данного синдрома необходимо записаться на прием к терапевту или педиатру. Также необходима консультация кардиолога и офтальмолога. Врач составит анамнез и проведет первичный опрос.
Исследования, при помощи которых можно диагностировать данное заболевание:
Арахнодактилия при гомоцистинурии
Гомоцистинурия наследуется по аутосомно-рецессивному типу. Данное заболевание и синдром Марфана почти идентичны по внешним проявлениям: при гомоцистинурии также наблюдается выраженная арахнодактилия, удлинение трубчатых костей,деформации грудной клетки, таза и позвоночника, нарушения со стороны сердечно-сосудистой системы и со стороны глаз. Наряду с этим, при гомоцистинурии арахнодактилия сочетается с нарушениями нервно-психической деятельности: снижением IQ до 32-72 единиц (в норме 85-115 единиц), низкой работоспособностью, проблемами при переключении внимания, некритичным отношением к своим возможностям, упрощенной речью, ослаблением мимики и т.д.
Каковы симптомы железодефицитной анемии у беременных?
Недостаток железа в организме проявляется большим разнообразием симптомов. А у будущих мам патологические признаки путаются с физиологическими проявлениями беременности.
Выделяют две большие группы признаков анемии — гипоксический синдром и сидеропенический синдром.
Симптомы, обусловленные кислородным голоданием:
слабость;
снижение работоспособности;
бледность кожных покровов и слизистых оболочек;
мелькание «мушек» перед глазами;
одышка при физической нагрузке;
головная боль;
головокружение;
шум в ушах;
усиленное сердцебиение;
тошнота;
нарушения сна;
апатия;
обмороки.
Признаки сидеропенического синдрома, обусловленного недостатком железа в тканях:
сухость и шелушение кожи;
ломкость и слоистость ногтей;
выпадение волос;
извращения вкуса — пристрастие к мелу, извести, глине;
токсикомания — тяга к запахам ацетона, лака, краски, бензина;
язвы и трещины в уголках рта;
жжение языка;
нарушение работы органов пищеварения: обострение хронического гастрита, синдромы нарушения всасывания в кишечнике, затрудненное глотание сухой и твердой пищи;
миокардиодистрофия, недостаточность мышечных сфинктеров и другие тяжелые проявления в запущенных случаях.
Многообразие клинических симптомов обусловлено тем, что железо входит в состав и контролирует работу большого количества ферментов. А его недостаток приводит к разладу в обменных процессах и, как следствие, к метаболическим нарушениям.







Другие заболевания из группы Болезни органов дыхания:
| Агенезия и Аплазия |
| Актиномикоз |
| Альвеококкоз |
| Альвеолярный протеиноз легких |
| Амебиаз |
| Артериальная легочная гипертония |
| Аскаридоз |
| Аспергиллез |
| Бензиновая пневмония |
| Бластомикоз североамериканский |
| Бронхиальная Астма |
| Бронхиальная астма у ребенка |
| Бронхиальные свищи |
| Бронхогенные кисты легкого |
| Бронхоэктатическая болезнь |
| Врожденная долевая эмфизема |
| Гамартома |
| Гидроторакс |
| Гистоплазмоз |
| Гранулематоз вегенера |
| Гуморальные формы иммунологической недостаточности |
| Добавочное легкое |
| Ехинококкоз |
| Идиопатический Гемосидероз легких |
| Идиопатический фиброзирующий альвеолит |
| Инфильтративный туберкулез легких |
| Кавернозный туберкулез легких |
| Кандидоз |
| Кандидоз легких (легочный кандидоз) |
| Кистонозная Гипоплазия |
| Кокцидиоилоз |
| Комбинированные формы иммунологической недостаточности |
| Кониотуберкулез |
| Криптококкоз |
| Ларингит |
| Легочный эозинофильный инфильтрат |
| Лейомиоматоз |
| Муковисцидоз |
| Мукороз |
| Нокардиоз (атипичный актиномикоз) |
| Обратное расположение легких |
| остеопластическая трахеобронхопатия |
| Острая пневмония |
| Острые респираторные заболевания |
| Острый абсцесс и гангрена легких |
| Острый бронхит |
| Острый милиарный туберкулез легких |
| Острый назофарингит (насморк) |
| Острый обструктивный ларингит (круп) |
| Острый тонзиллит (ангина) |
| Очаговый туберкулез легких |
| Парагонимоз |
| Первичный бронхолегочный амилоидоз |
| Первичный туберкулезный комплекс |
| Плевриты |
| Пневмокониозы |
| Пневмосклероз |
| Пневмоцитоз |
| Подострый диссеминированный туберкулез легких |
| поражение газами промышленного происхождения |
| Поражение легких вследствие побочного действия лекарственных препаратов |
| поражение легких при диффузных болезнях соединительной ткани |
| Поражение легких при болезнях крови |
| Поражение легких при гистиоцитозе |
| Поражение легких при дефеците а 1- антитрипсина |
| поражение легких при лимфогранулематозе |
| Поражение легких при синдроме Стивенса-Джононса |
| Поражения легких отравляющими веществами |
| Пороки развития легких |
| Простая Гипоплазия |
| Радиационные поражения легких |
| Рецидивирующий бронхит у детей |
| Саркаидоз органов дыхания |
| Секвестрация легкого |
| Синдром гудпасчера |
| Синдром Маклеода |
| Синдром Мендельсона |
| Синусит |
| Спонтанный пневмоторакс |
| Споротрихоз |
| Стафилококковые деструкции легких у детей |
| Стенозы и трахеи крупных бронхов |
| Стенозы и трахеи крупных бронхов |
| Стрептококковый фарингит |
| Сфеноидальный синусит (сфеноидит) |
| Токсоплазмоз |
| Трахеальный бронх |
| Трахеит |
| Трахеобронхомегалия |
| Тромбоэмболия легочной артерии (ТЭЛА) |
| Туберкулез внутригрудных лимфатических узлов (бронхоадениты) |
| Туберкулез бронхов, трахеи, верхних дыхательных путей |
| Туберкулез гортани |
| Туберкулез легких |
| Туберкулез полости рта, миндалин и языка |
| Туберкулезная интоксикация у детей и подростков |
| Туберкулезный плеврит |
| Туберкулема легких |
| Фарингит |
| Фиброзно-кавернозный туберкулез |
| Фронтит (острый фронтальный синусит) |
| Хроническая пневмония |
| Хроническая пневмония у детей |
| Хронический абсцесс легких |
| Хронический бронхит |
| Хронический гематогенно-диссеминированный туберкулез легких |
| Хроническое легочное сердце |
| Цирротический туберкулез легких |
| Шистосомозы |
| Экзогенный аллергический альвеолит |
| Эмфизема легких |
| Эпиглоттит |
| Этмоидальный синусит (этмоидит) |
Диагностика
Диагноз синдром Марфана ставится на основании имеющейся клинической картины, семейного анамнеза и генетических исследований.
Процессу диагностики помогает ряд визуальных исследований. Рентгенография дает информацию о состоянии грудной клетки, изменении длины и ширины костей ладоней и пальцев. Рентгенограммы таза отражают состояние тазобедренных суставов. КТ и МРТ имеют преимущества в оценке состояния мягких тканей и детализации дефектов.
Сердечно-сосудистая система оценивается на основании физического осмотра, касающегося дефекта клапана, признаков сердечной недостаточности, ЭКГ и обычной, а в некоторых случаях чреспищеводной эхокардиографии. Состояние аорты и крупных сосудов оценивают контрастным исследованием.
Исследование зрительной системы начинается с определения остроты зрения, офтальмоскопии, осмотра глазного дна. Широко распространенным исследованием в последнее время является УЗИ глазного яблока.
Заключение
Прогноз и течение синдрома Марфана зависит от выраженности поражения гена и недостаточного синтеза белка фибрина.
До 90% больных синдромом Марфана не доживают до 50 лет.
При отсутствии должной и регулярной диагностики у пациента резко повышается риск внезапной смерти. Новые технологии в современной медицине могут существенно продлить жизнь человека и улучшить её качество.
Анастасия Минина, Врач-педиатр 40 статей на сайте
После окончания медицинского ВУЗа я прошла подготовку в интернатуре по направлению: «Поликлиническая педиатрия». После прохождения обучения я работаю районным педиатром Холмогорской больницы села Холмогоры Архангельской области.





