Причины генетической патологии
ДНКсовокупность геновферментовВсе хромосомы можно условно разделить на два вида:
- Аутосомы. Аутосомы – это первые 22 пары хромосом. Они кодируют большую часть генетической информации. Каждая из аутосом содержит гены, без которых организм попросту не может выжить. Именно поэтому отсутствие любой из 44 аутосом неминуемо ведет к гибели плода во внутриутробном периоде.
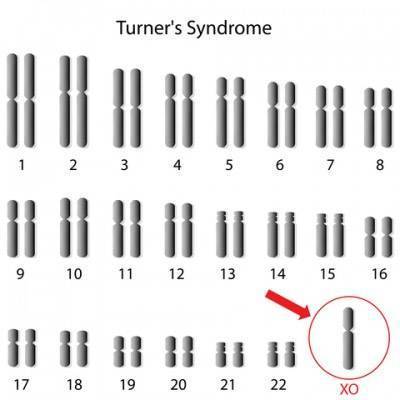
- Половые хромосомы. Половые хромосомы, в отличие от аутосом, не всегда являются схожими, хотя их тоже условно объединяют в 23-ю пару. Эти хромосомы бывают двух видов, которые условно обозначаются Х (женская хромосома) и Y (мужская хромосома). Эти молекулы ДНК сильно различаются между собой как по размерам, так и по генам, которые на них расположены. Хромосома Y является самой маленькой в геноме и содержит всего 429 генов (из них лишь 76 кодируют определенные белковые молекулы). Присутствие этой хромосомы в геноме необязательно. Она обуславливает развитие организма по мужскому типу. Если же ее нет, идет развитие по женскому типу.
беременностьСуществуют следующие генетические варианты данного заболевания:
- Полное отсутствие второй половой хромосомы. Данный вариант синдрома встречается чаще всего (примерно в 60% случаев). Единственная Х-хромосома почти в 80% случаев имеет материнское происхождение. Поскольку вторая (отцовская) хромосома отсутствует, развитие по женскому типу оказывается неполным. До 3 месяца беременности яичники у эмбриона развиваются нормально. Однако дальше происходит постепенное замещение нормальных фолликулов (клетки, из которых в будущем сформируются яйцеклетки) соединительной тканью. Кроме того, наблюдается ряд пороков развития других органов и тканей. Полное отсутствие второй половой хромосомы считается наиболее тяжелым вариантом синдрома. При нем основные признаки и симптомы встречаются наиболее часто.
- Мозаичный вариант. Мозаичный вариант считается более легким. При нем только часть клеток организма содержит одну Х-хромосому. В других клетках половых хромосом две, причем вторая может быть как Х, так и Y. Иногда встречаются и больные с третьим типом клеток, содержащим три Х-хромосомы. Механизм образования такой мозаики достаточно прост. После зачатия клетки эмбриона начинают активно делиться. При этом делится и генетический материал между дочерними клетками. Если в какой-то момент этого разделения не происходит, то часть клеток в будущем будет иметь аномальный набор половых хромосом. При мозаичном варианте прогноз для девочек лучше. Тяжелые пороки развития при рождении обычно отсутствуют, а в пубертатном периоде проблемы с менструальным циклом не так сильно выражены. Как правило, фенотип (внешность), характерная для синдрома Шерешевского-Тёрнера все же присутствует, но количество видимых признаков этого заболевания меньше, чем при полной моносомии Х.
- Структурные изменения Х-хромосомы. В редких случаях синдром Шерешевского-Тёрнера диагностируют у женщин, которые по факту имеют две Х-хромосомы. Одна из этих хромосом сильно повреждена (например, отсутствует ее большая часть). При этом появляются симптомы и признаки, характерные для этого заболевания, но их будет меньше, чем при полной форме.
для женщин – 46, ХХНа вероятность рождения ребенка с синдромом Шерешевского-Тёрнера могут повлиять:
- некоторые инфекции половой сферы, перенесенные женщиной в прошлом;
- химические факторы (загрязненность окружающей среды, сложные химические соединения);
- физические факторы (сильные электромагнитные и ионизирующие излучения);
- генетическая предрасположенность;
- голодание или истощение организма в результате тяжелой болезни (преимущественно в период перед зачатием).
гаметы
Установление диагноза
У младенца синдром Шерешевского может выявить неонатолог или педиатр, для этого достаточно провести визуальный осмотр. Указывает на наличие патологии крыловидные складки на шее и отечные конечности. Если явные наружные симптомы отсутствуют, то болезнь обнаруживается в подростковом возрасте по низкому росту, отсутствию первого менструального кровотечения. Кроме того, у больного слабо выражены вторичные половые признаки.
Диагностика обязательно включает анализ на гормоны. Во время исследования обнаруживается увеличение количества гонадотропинов, снижение уровня эстрогенов. Большое диагностическое значение имеет исследование и кариотипа. Обнаружить специфические признаки патологии врач может после акушерского УЗИ. При подозрении на СШТ назначается пренатальная диагностика:
- УЗИ плода показывает, что у него отсутствует носовая кость, увеличена толщина воротникового пространства, недостаточно длинные бедренные или плечевые кости и т. д. Комплексное исследование включает биохимию крови матери на β-ХГЧ, РАРР-А.
- Инвазивные исследования (амниоцентез, биопсия хориона) являются более точными, чем вышеописанный скрининг. Пункцию амниотической оболочки (амниоцентез) и биопсию хориона (зародышевая часть плаценты) чаще всего назначают при повышенном риске рождения ребенка с СШТ, женщинам старше 35 лет, а также при плохих результатах неинвазивных исследований.
Инвазивные исследования проводят только по особым показаниям, они способны с высокой точностью определить патологию.
Больному может понадобиться помощь различных специалистов: генетика, эндокринолога, кардиолога, нефролога, офтальмолога, ЛОР-врача, гинеколога, андролога и т. д.
Чтобы обнаружить врожденные пороки и сопутствующие болезни, назначаются следующие исследования:
- Эхокардиография.
- МРТ сердца.
- Электрокардиограмма.
- УЗИ почек.
- Рентген позвоночного столба, суставов конечностей и остального скелета.
- Денситометрия для проверки плотности костей.
- Гинекологические исследования.
- УЗИ органов малого таза, мошонки и т. д.
Чтобы отличить СШТ от гипофизарного нанизма (задержка физического развития вследствие недостатка соматотропного гормона), нужно провести анализ на гормоны гипофиза, рентген турецкого седла (образование в клиновидной кости черепа), ЭЭГ (электроэнцефалографию).
Мозаичная форма Шерешевского-Тернера
Считается, что мозаичная форма Шершевского-Тернера имеет более легкое течение в отличие от классической патологии. Главная ее особенность – это частичное развитие патологии в клетках. То есть часть клеток организма имеет нормальный кариотип, а часть – аномальный. По этой причине симптоматика менее выраженная, чем при классической форме патологии.
Формы СШТ в зависимости от характера повреждения второй Х-хромосомы (по кариотипу):
- 46 Х Хр и 46 Х Хq.
- 46 Xi(Xq) 46,X,i(XP).
- 4 X/46 XY.
Как видно, мозаичный вариант патологии почти во всех случаях имеет нормальное количество хромосом. Однако наблюдаются некоторые дефекты в плечах одной из них. Именно это приводит к появлению симптомов заболевания. Они возникают в основном из-за нарушения процесса синтеза белков, что приводит к порокам развития.
Этиопатогенетические факторы
Синдром Шершевского-Тернера — наследственная патология, обусловленная отсутствием половой хромосомы, которая, можно сказать, окончательно делает из женщины женщину. Некорректное деление клеток в процессе зачатия приводит к выстраиванию генетического материала с явными нарушениями. К указанному хромосомному дисбалансу ведут неправильно «вставшие» молекулы ДНК.
Недуг также развивается в результате неправильного формирования Х-хромосомы. Причинами ее аномального строения являются:
- потеря участка хромосомы в результате ее разрыва,
- перенес участка хромосомы,
- образование хромосомы в виде кольца,
- прочие хромосомные перестройки — мутации или аберрации.
Мозаицизм имеет большое значение в развитии недуга. У больных в тканях обнаруживают генетически разнородные клетки в различных вариациях. Все эти варианты характерны для женщин. У мужчин синдром возникает крайне редко. Его основными причинами являются транслокация или мозаицизм. Мейотическое расхождение хромосом лежит в основе патологического процесса.
Некорректная хромосомная «сборка» может заключаться в наличии Y-хромосомного элемента в кариотипе. Таким больным удаляют яичники. Это необходимое мероприятие, позволяющее продлить жизнь людям с данным недугом, поскольку Y-хромосомный элемент часто провоцирует развитие раковой опухоли — гонадобластомы.
Кариотипы при данном синдроме:
- Кариотип 45Х0 — замена железистой ткани яичника соединительнотканными тяжами. Нефункционирующие женские железы приводят к необратимому бесплодию. Для продолжения рода прибегают к помощи ЭКО. Этот тип синдрома является самым распространенным и одним из самых тяжелых. Он отличается ярко выраженной симптоматикой и развитием тяжелых осложнений. Синдром с трудом поддается лечению.
- Мозаичный кариотип 45 X0/46 XY – отсутствие матки и недоразвитие влагалища, высокий риск онкологии. Для предупреждения рецидивов болезни показано удаление яичников. Мозаичный кариотип 45 Х0/46 ХХ — ничтожно малые размеры яичников. Беременность возможна при участии донорской яйцеклетки. Мозаичный тип синдрома отличается более легким течением: пороки развития не наблюдаются, симптомов возникает намного меньше и они менее выражены. Недуг хорошо лечится. Мозаичный тип отличается сочетанием двух видов клеток — с нормальным кариотипом и без одной Х-хромосомы. От их пропорционального соотношения будет зависеть состояние здоровья женщины.
Первоначально у эмбриона закладывает нормальное количество половых клеток. В процессе роста и развития плода они подвергаются инволюции. Новорожденная девочка имеет очень мало фолликулов в яичнике или не имеет их вовсе. Кроме дисфункции яичников у больных в процессе эмбриогенеза формируются многочисленные пороки внутренних органов.





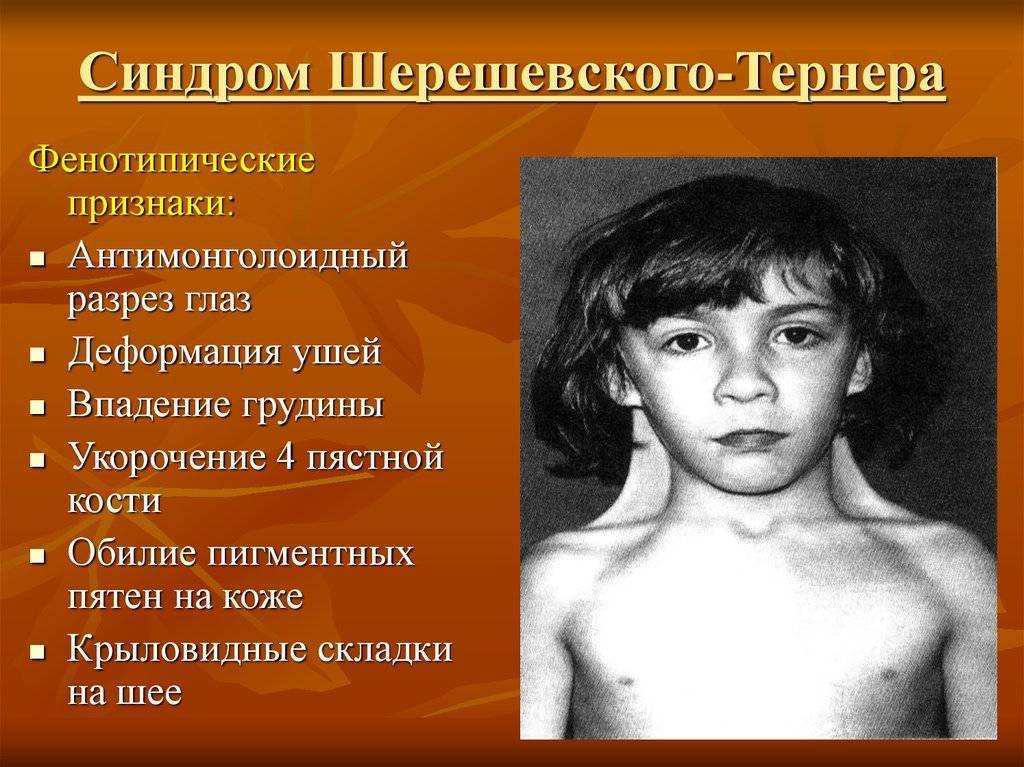
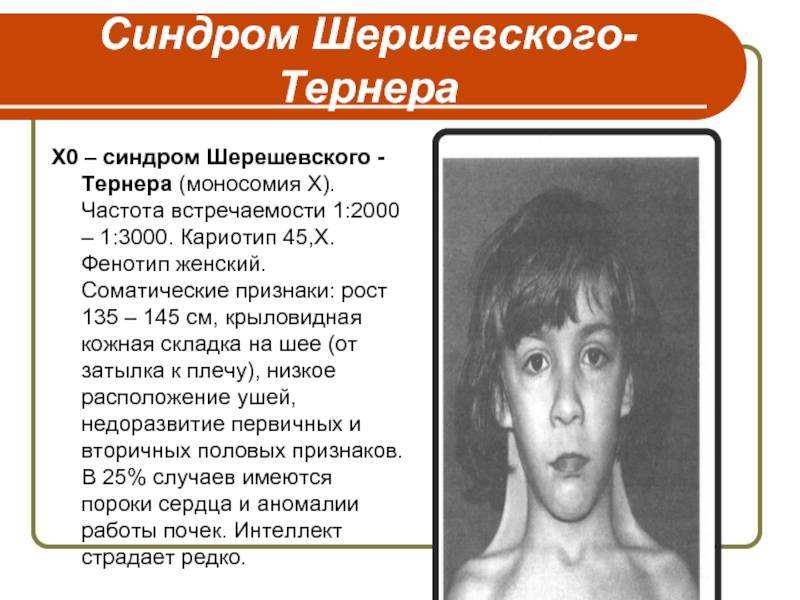
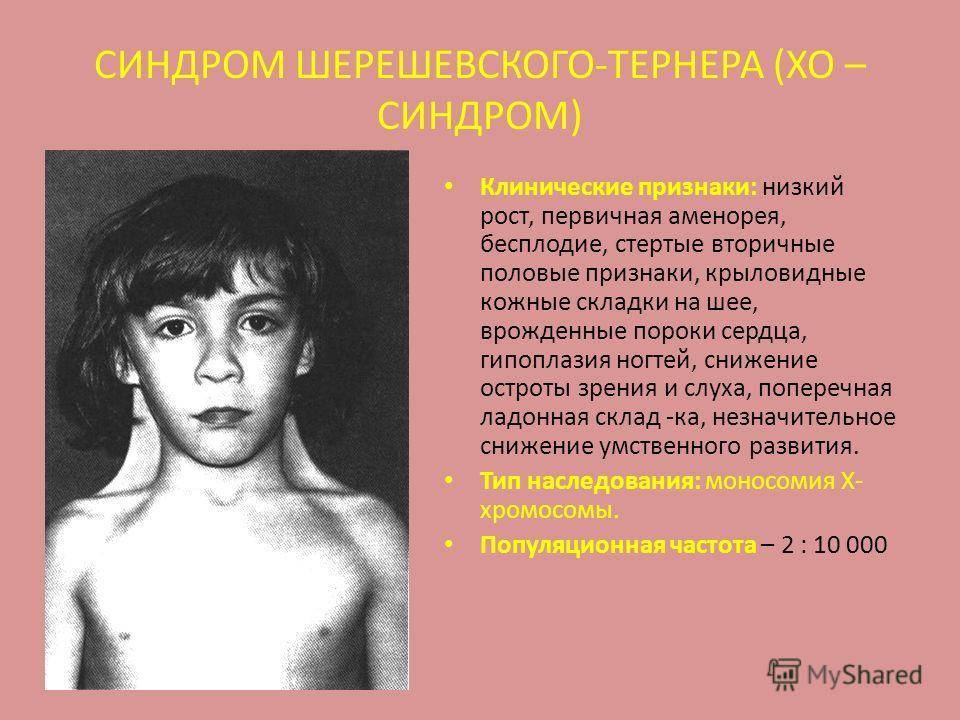
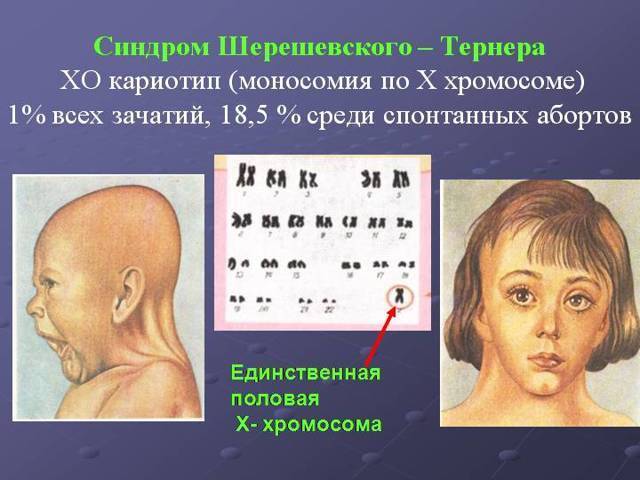
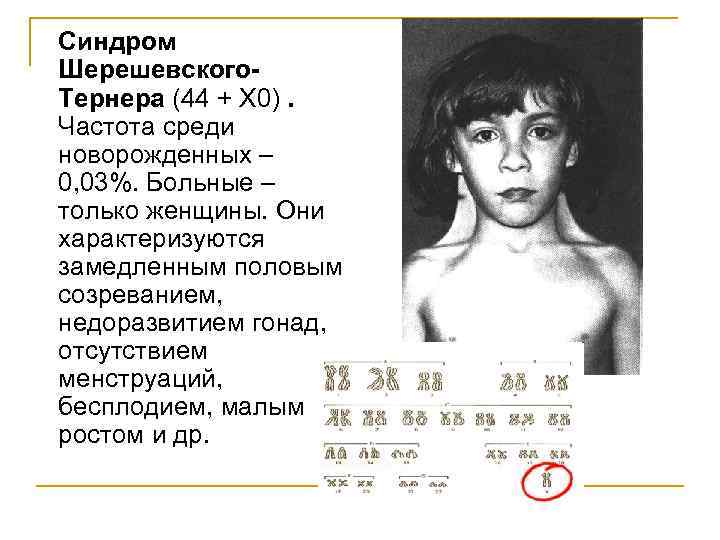
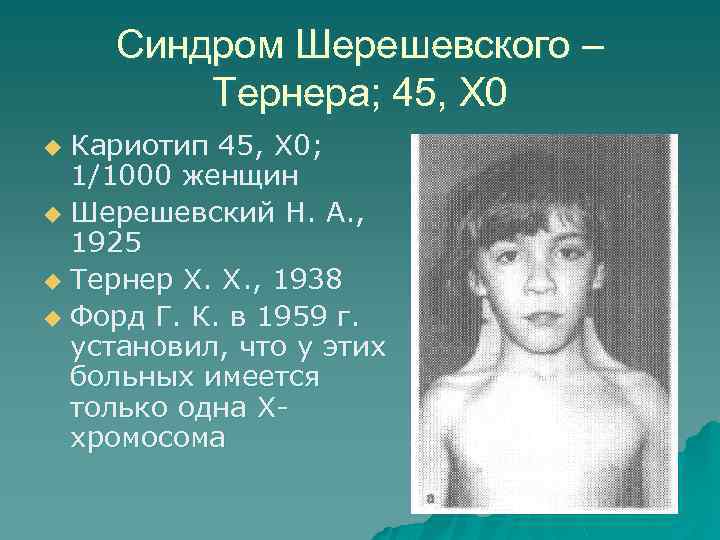
Синдром Шерешевского-Тернера
Синдром Шерешевского—Тернера
Синдром впервые
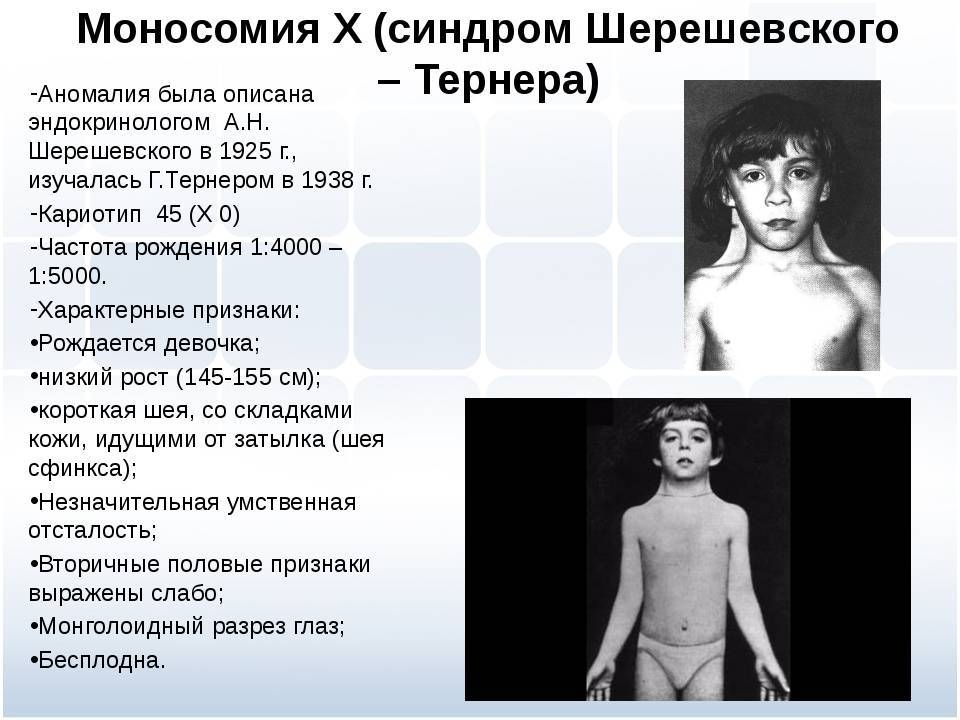
описан Н. Шерешевским в 1925 г., а затем Г. Тернером в 1938 г. В 1959 г. Форд установил, что у больных с этим синдромом отсутствует одна Х-хромосома. Болеют
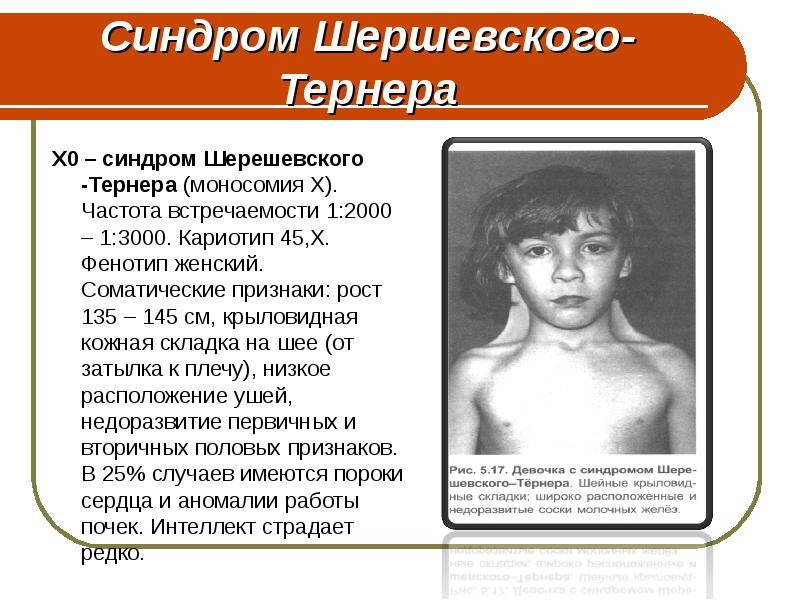
почти исключительно женщины. Их кариотип 45, ХО. Заболевание обычно выявляется
в пубертатном периоде, когда у девочек отмечаются признаки полового
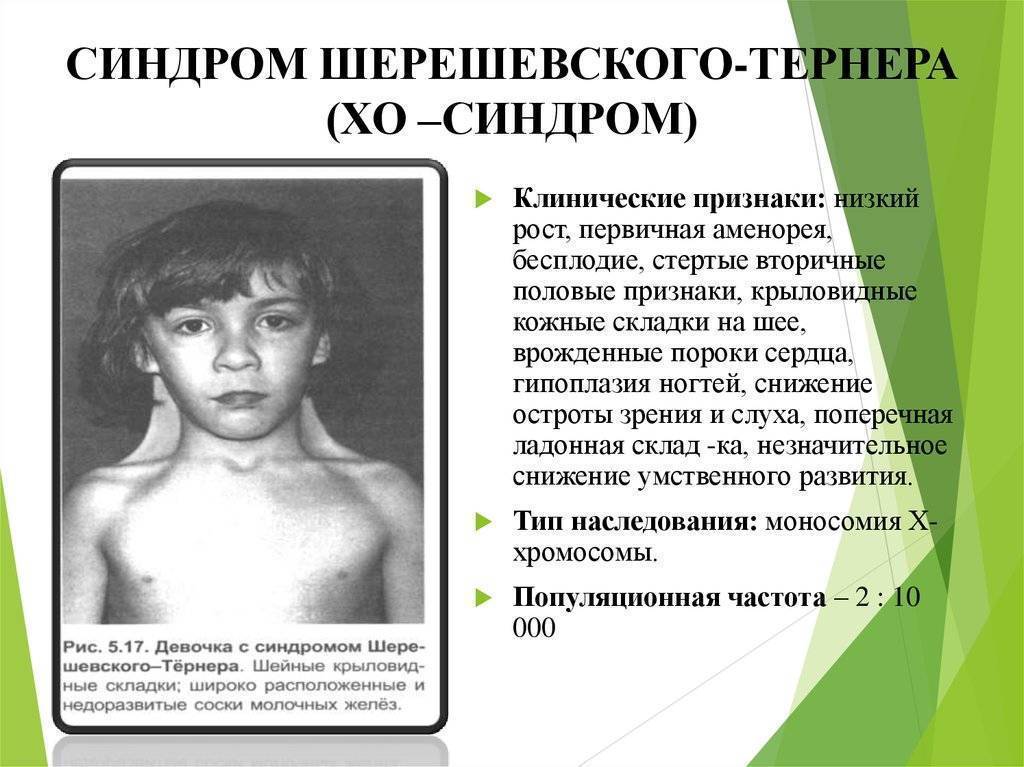
инфантилизма. Больные низкого роста, с короткой шеей, по бокам которой
отмечаются кожно-мышечные складки, идущие от затылка к надплечиям. Ушные
раковины деформированы и низко расположены. Волосы на шее растут низко. Нередко
отмечаются микрогнатия, ретрогнатия, эпикант. Грудные железы обычно
отсутствуют. На их месте определяется складка жира. Соски недоразвиты, ареолы
втянуты, широко расположены и не пигментированы. Наружные половые органы недоразвиты,
влагалище узкое, клитор гипертрофирован, матка и яичники недоразвиты. Месячные
отсутствуют или бывают однократными. Больные бесплодны Отмечаются изменения и
со стороны конечностей: вальгусное положение локтевых суставов, широкие
короткие ладони, укорочение безымянного пальца, деформация мизинца. Ногти
глубоко посажены и деформированы, на кончиках пальцев преобладают круговые
узоры, из-за чего увеличен дельтовидный индекс и гребневый счет. Угол a td
увеличен и приближается к прямому.
Ноги укороченные,
толстые. 3, 4-й и 5-й пальцы укорочены, искривлены и неправильно располагаются
над стопой. На рентгенограммах трубчатых костей отмечается задержка
окостенения, хотя рост таких женщин прекращается в 15—18 лет. Определяется
увеличение медиальных мыщелков бедренных костей и уменьшение латеральных,
истончение латеральных концов ключицы, слияние тел позвонков, сужение ребер,
остеопороз костей, особенно метафизарных частей трубчатых костей.
Нередко
обнаруживаются пороки развития сердечно-сосудистой системы, изменения со
стороны внутренних органов (коаркта-ция аорты, ее атрезия, стеноз легочной
артерии, незаращение межжелудочковой перегородки, подковообразная почка и др.).
Со стороны нервной системы существенных изменений не отмечается. Интеллект этих
больных страдает мало или вообще не нарушен.
Этиология синдрома
Шерешевского—Тернера не ясна. Возраст родителей таких детей значения не имеет;
они обычно низкого роста, хотя кариотип их нормальный.
Окончательный
диагноз устанавливается на основании исследования кариотипа больных. Важным в
диагностике синдрома Шерешевского—Тернера является отсутствие полового
хроматина в клетках букального эпителия,
Известно, что в
соматических клетках женского организма одна Х-хромосома в интерфазе не
активна, она спирализована и образует половой хроматин, который обнаруживается
у ядерной оболочки. В клетках мужского организма полового хроматина нет, так
как у них только одна Х-хромосома и она функционирует в интерфазе. В
сегментоядерных лейкоцитах периферической крови женщин обнаруживаются
палочкообразные выпячивания ядер — барабанные палочки или тельца Барра, которые
трактуются как спирализованные Х-хромосомы. В крови здоровых мужчин и у женщин
с синдромом Шерешевского—Тернера барабанные палочки не обнаруживаются. Таким
образом, на основании обнаружения полового хроматина в клетках слизистой щеки и
барабанных палочек в лейкоцитах периферической крови можно предварительно
поставить диагноз синдрома Шерешевского—Тернера, но
окончательно диагноз устанавливается на основании исследования кариотипа, где
обнаруживается 45 хромосом (45, ХО).
Описаны случаи
синдрома Шерешевского—Тернера и у мужчин с нормальным кариотипом 46, XY. Такие
мужчины низкого роста, с короткой шеей, бочкообразной грудной клеткой,
вальгус-ной установкой локтевых суставов, гипоплазией нижней челюсти, высоким
небом, недоразвитием тестикул, стенозом легочной артерии и умственной
отсталостью. При гистологическом исследовании гонад отмечается малое число половых
клеток, поэтому эти больные, как правило, бесплодны. Несмотря на нормальный
кариотип — 46, XY, у них Y-хромосома не активна и фактически генотип
соответствует ХО.
Описаны варианты
синдрома Шерешевского—Тернера с мозаи-цизмом, когда одна часть клеток имеет
кариотип 45, ХО, а другая — нормальный кариотип 46, XX. Мозаичные варианты
синдрома протекают мягче. У таких женщин возможны менструации, беременность,
роды.
Источник информации:
Интернет:
Можно ли делать ЭКО?
Синдром Тернера сопровождается бесплодностью. Возможность иметь детей есть только у пациенток с мозаичной формой, у которых сформирована матка. Половой инфантилизм не позволяет провести зачатие естественным путем. Шанс выносить и родить здорового ребенка есть у женщины при проведении ЭКО.
Если у женщины полноценно развиты яичника, ей подсаживают собственную оплодотворенную яйцеклетку. Чтобы ребенок родился без нарушений на генном уровне, половые клетки должны иметь нормальный кариотип 46ХХ. Современные репродуктивные методы позволяют использовать донорскую яйцеклетку.
Успешное оплодотворение происходит в 2–3 раза реже, чем у здоровых женщин, и составляет 20–30%. Вероятность прерывания беременности после ЭКО составляет у больных пациенток до 40%.
Как наблюдают девочек с аномалией Х
Поскольку моносомия Х связана с изменением в генетическом материале, эта болезнь неизлечима. Пациента ставят на учет сначала к педиатру, потом к терапевту, а также к нефрологу, кардиологу, эндокринологу, отоларингологу, стоматологу и другим врачам по показаниям.
В первый год жизни необходимо обследовать зрительную систему. У девочек до 4 лет проверяют состояние тазобедренных суставов, чтобы исключить дисплазию. Нередко требуется коррекция сколиоза, а после 18 лет лечение остеопороза. Оценку функциональности сердца проводят каждые 5 лет (ЭКГ, ЭхоКГ, МРТ). С такой же периодичностью проверяют органы слуха.
До 18 лет необходимо каждый год проверять уровень гормонов щитовидной железы, работу почек и печени
В будущем важно также контролировать уровень холестерина и сахара в крови. Каждые 3 года следует сдавать анализ на антитела к трансглутаминазе, чтобы вовремя диагностировать целиакию
Этиология и патогенез
В 50—60-х гг. Фордом (С. E. Ford) и др. у большинства обследованных ими больных с Тернера синдромом была обнаружена гоносомная моносомия 45,X, т. е. отсутствие одной из половых хромосом — X или Y (см. Хромосомный набор). Позднее были выявлены варианты Т. с. со структурными дефектами X-хромосомы (делеция короткого плеча, изо-Х-хромосома по длинному плечу, кольцевая X-хромосома и др.) и с хромосомным мозаицизмом 45,Х/46, XX; 45,Х/46,XY; 45,Х/46,ХХ/47,ХХХ и др. (см. Мозаицизм). Соответственно кариотипу (см.) у большинства больных с Т. с. половой хроматин (см.) отсутствует, при мозаичных вариантах Т. с. с наличием в части клеток нормального женского кариотипа 46,XX содержание полового хроматина снижено, при структурных дефектах Х-хромосомы размеры Х-хроматина (телец Барра) могут быть увеличены или уменьшены.



Четкой связи возникновения Т. с. с возрастом и какими-либо заболеваниями родителей не выявлено. Однако беременности, заканчивающиеся рождением детей с Т. с., нередко бывают осложненными токсикозами беременных (см.), угрозой выкидыша. Роды (см.) часто бывают преждевременными и патологическими. Особенности беременностей и родов, заканчивающихся рождением ребенка с Т. с., вероятно, следствие хромосомной патологии плода.
Нарушение формирования половых желез при Т. с. обусловлено отсутствием или структурными дефектами одной половой хромосомы. Установлено, что в таком случае у эмбриона первичные герминативные клетки закладываются почти в нормальном количестве, однако во втором триместре беременности происходит их быстрая инволюция и к моменту рождения ребенка количество примордиальных фолликулов в яичнике по сравнению с нормой резко уменьшено или они полностью отсутствуют. Это приводит к выраженной эстрогенной недостаточности, половому недоразвитию, у большинства больных — к первичной аменорее (см.) и бесплодию (см.).
Хромосомный дисбаланс в результате отсутствия одной из половых хромосом или части X-хромосомы является причиной возникновения различных дефектов соматического развития. Возможно также, что сопутствующие аутосомные мутации играют определенную роль в появлении пороков развития, поскольку существуют состояния, соматически сходные с Т. с., но без видимой хромосомной патологии и без полового недоразвития (синдром Нунен, отдельные случаи Т. с. у мужчин),
Отсутствие X-хромосомы приводит к проявлению у ряда больных с Т. с. рецессивных генов, расположенных в Х-хромосоме, что является причиной дальтонизма (см. Цветовое зрение) у таких больных (мужчин и женщин), недостаточности глюкозо-6-фосфатдегидрогеназы (КФ 1.1.1.49) и др. и определяется частотой соответствующих мутантных генов в популяции.
Диагностика синдрома Шерешевского-Тёрнера
У новорожденных диагноз можно заподозрить при наличии лимфедемы или крыловидной складки шеи. В отсутствие этих изменений диагноз у некоторых детей выявляют позже на основании низкого роста, аменореи и отсутствия пубертата. Диагноз подтверждается исследованием кариотипа. Для обнаружения врожденных пороков сердца показано проведение эхокардиографии или МРТ.
Цитогенетический анализ и исследования с Y-специфичным зондом проводят всем лицам с дисгенезией гонад для исключения мозаицизма с наличием клеточной линии с кариотипом 46, XY (45, X/46XY). У таких пациентов обычно женский фенотип с различными чертами синдрома Тернера. Они находятся в группе повышенного риска по развитию злокачественных новообразований гонад, особенно гонадобластомы, поэтому для профилактики сразу после определения диагноза следует удалить гонады.
Физическое обследование
Диагноз ставят на основании характерной клинической картины: короткая шея с избытком кожи и крыловидными складками у новорождённых девочек, лимфатический отёк кистей и/или стоп, врождённые пороки левого сердца или аорты (особенно коарктация аорты), задержка роста и полового развития в пубертатный период у девочек.
Лечение синдрома Шерешевского – Тернера в Израиле
Лечение данного заболевания направлено на устранение тех симптомов, которые вызываются аномалией набора хромосом.
- В детстве проводится терапия препаратами, целью и функцией которых является стимуляция роста пациентов. Для этого применяются анаболические стероиды. Однако в таком случае необходимо помнить об их побочных эффектах и осуществлять контроль со стороны гинеколога.
- Также необходимо проводить терапию эстрогенами, что позволяет сделать больную более женственной и устранить некоторые признаки заболевания. Однако эта терапия проводится только с 14-16 лет, когда половые гормоны начинают вырабатываться в норме. Также раннее начало терапии эстрогенами недопустимо из-за того, что они могут вызвать закрытие зон роста в костях и низкорослости.
- Гормональная терапия проводится на протяжении длительного времени. Иногда под действием гормонов развиваются вполне нормальные репродуктивные органы. Это позволяет при использовании донорской яйцеклетки и ЭКО забеременеть и родить вполне здорового ребенка.
Если заболевание не сопровождается серьезными аномалиями со стороны жизненно важных органов (преимущественно сердца), то прогноз достаточно благоприятный. А при проведении адекватной терапии качество жизни пациентов находится на вполне достойном уровне.
Извините. Эта форма больше не принимает новые данные.
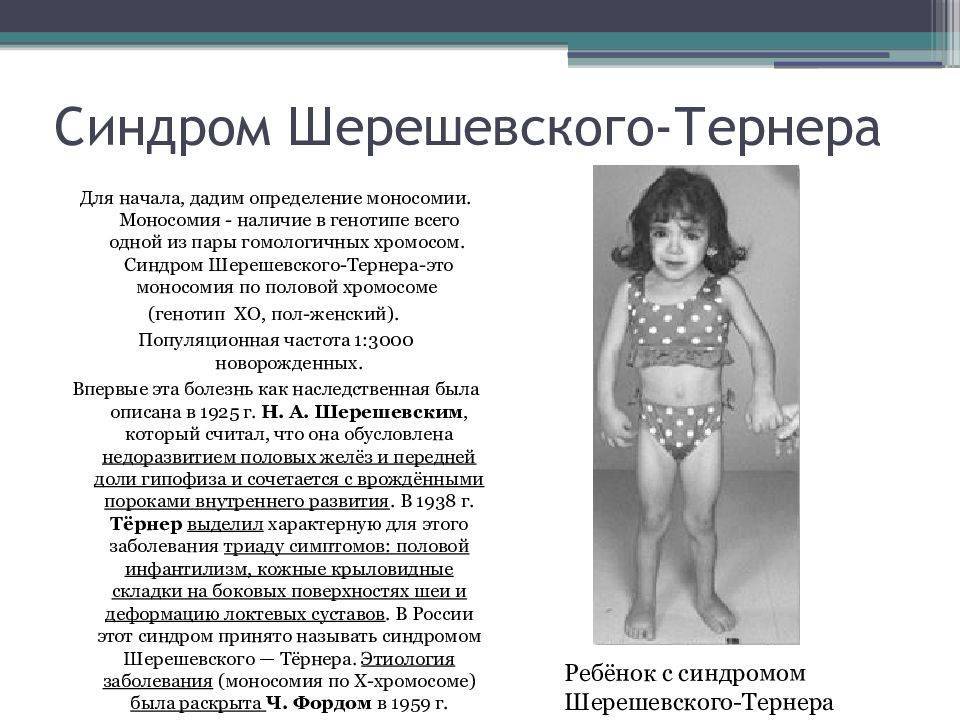
Причины синдрома Шерешевского-Тёрнера
У девочки с синдромом Тернера только 1 нормальная половая Х-хромосома, а не обычные 2. Все люди рождаются с 23 парами хромосом. Одна пара хромосом, половые хромосомы, определяет пол ребенка. 1 половая хромосома принадлежит отцу, а 1 – от матери. Вклад матери всегда – Х-хромосома. Вклад отца может быть либо X-, либо Y-хромосомой. У девочки обычно 2 X-хромосомы (XX), а у мальчиков – X- и Y-хромосомы (XY). У женщины с синдромом отсутствует часть или вся 1 половая хромосома. Это означает, что у нее всего 1 полная Х-хромосома. Y-хромосома определяет «мужественность», поэтому, если она отсутствует, как при синдроме Тернера, пол ребенка неизменно будет женским. Это хромосомное изменение происходит случайно, когда ребенок зачат в утробе матери.
Клинические проявления синдрома Мея-Тёрнера
К сожалению, синдром недостаточно хорошо изучен, поскольку является редким. Специалисты выделяют три стадии его развития:
- Первая – клинические проявления отсутствуют;
- Вторая – просвет подвздошной вены сужается, появляются симптомы венозной недостаточности;
- Третья – развиваются тромбозы.
Симптоматика синдрома – следующая:
- Отёчность левой ноги, нередко – с изменением цвета кожного покрова на синюшний или багровый;
- Интенсивная болевая симптоматика в поражённой области, которую невозможно устранить обезболивающими средствами;
- Заметные невооружённым глазом варикозные узлы на бедре, яичке (у мужчин) или половых губах (у женщин) с поражённой стороны;
- Усиление неприятных ощущений после физических нагрузок, в том числе и несущественных;
- Появление геморроидальных узлов.
И у мужчин и женщин, страдающих от синдрома, возникают проблемы с сексуальными контактами. Так, в первом случае появляются боли тянущего характера в мошонке, которые иррадиируют в мочеиспускательный канал и головку члена и становятся более интенсивными после полового акта. Во втором случае сексуальные контакты становятся и вовсе невозможными из-за болезненности узла.
Синдром Шерешевского – Тернера – признаки заболевания
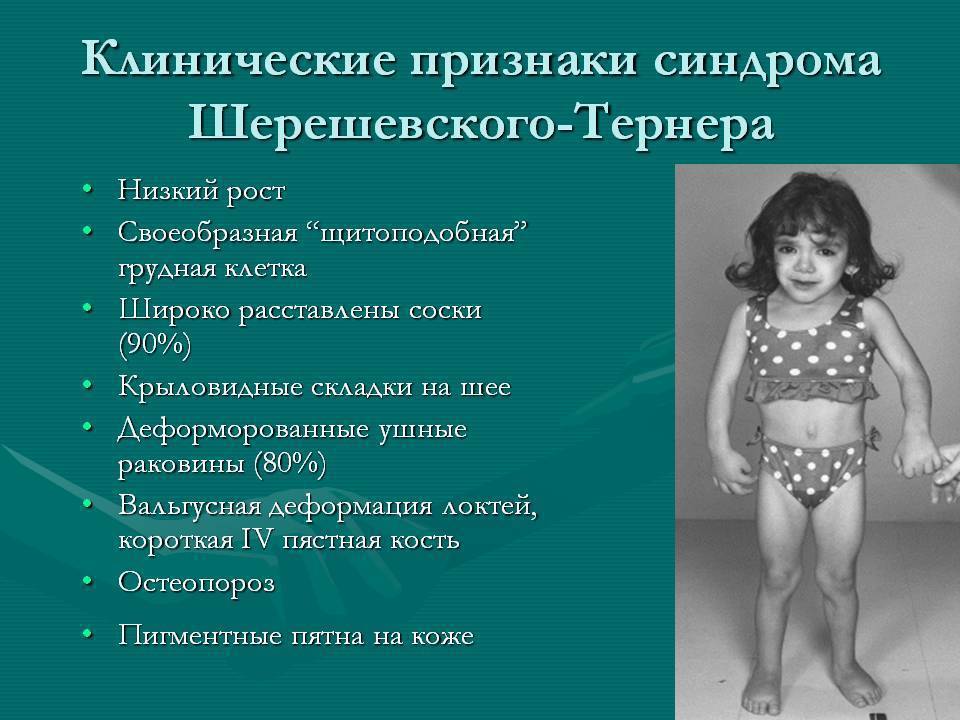
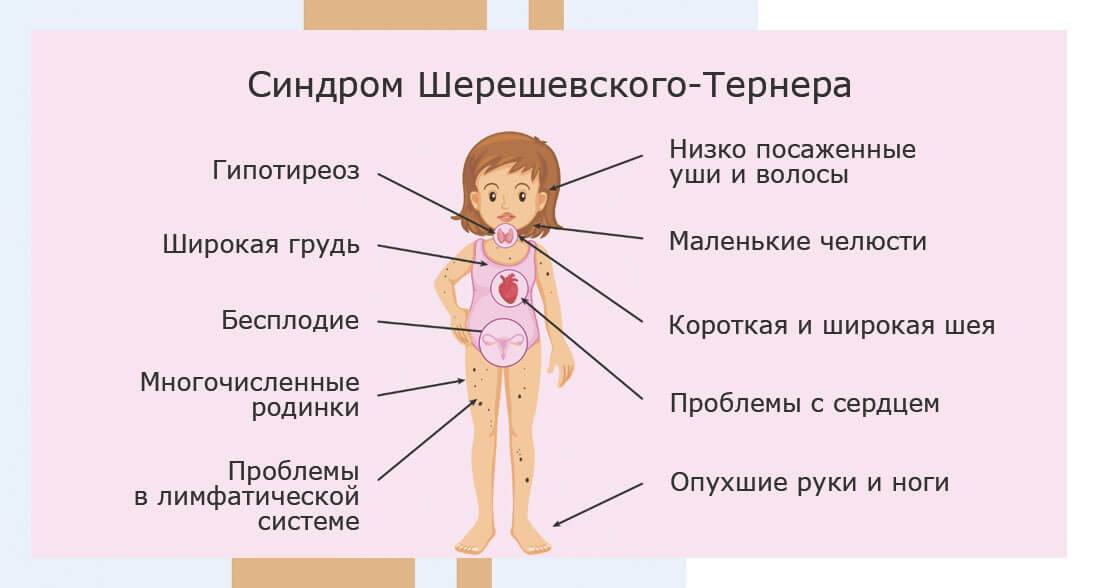
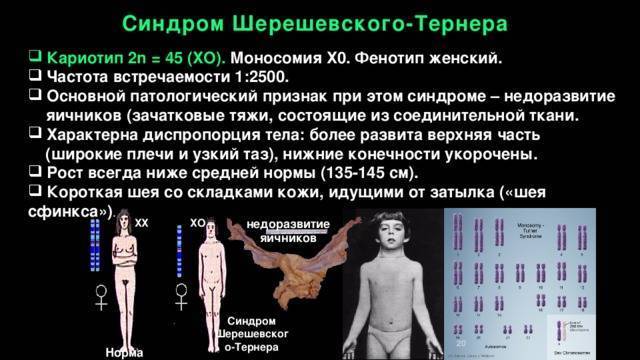
Больные с синдромом Шерешевского – Тернера предъявляют жалобы на отсутствие менструальных выделений, низкий рост и некоторые косметические дефекты внешности.
У большинства новорожденных девочек с этим заболеванием выявляются лишь легкие клинические проявления, однако у определенной группы младенцев отмечаются лимфатические отеки кистей и ступней, а также кожные складки на задней поверхности шеи. Одними из самых распространенных аномалий, встречающихся при заболевании Шерешевского – Тернера, считаются короткая шея с низким уровнем роста волос и крыловидными шейными складками, расширенная грудная клетка, слабо развитые половые органы – малые половые губы, клитор, матка, а также широко расставленные втянутые соски. У некоторых пациенток отмечаются аномалии формы и расположения ушей (лопоухость), которые сочетаются в половине случаев с тугоухостью.
В сравнении с родственниками девочки отличаются более низким ростом – не более 150 см, но при этом масса тела довольно внушительная. Реже выявляются такие признаки, как множественные пигментные невусы или витилиго, аномалии пястных и плюсневых костей кистей и стоп, деформация локтевых и плечевых суставов, гипоплазированные узкие ногти. Видоизменена и лицевая часть пациенток – челюстные кости уменьшены в размерах, небо высокое, наблюдается также опущение век.
Со стороны внутренних органов возможно обнаружение аномалий сердца в виде коарктации аорты, её расслоения, нарушения целостности межжелудочковой перегородки и двухстворчатого клапана аорты. Нередки и врожденные аномалии и пороки развития почек (подковообразная почка, удвоение лоханок и мочеточников), кровеносных сосудов опухолевой природы (гемангиомы, телеангиэктазии).
Женские половые железы замещаются тяжами из соединительной ткани, не способными вырабатывать половые клетки, в результате чего у подавляющего большинства (90%) больных девочек не наступает период полового созревания, молочные железы не увеличиваются, развивается первичная аменорея. У остальных 10% возможно спонтанное начало менструаций, но и в этом случае фертильность женщины остается под вопросом.
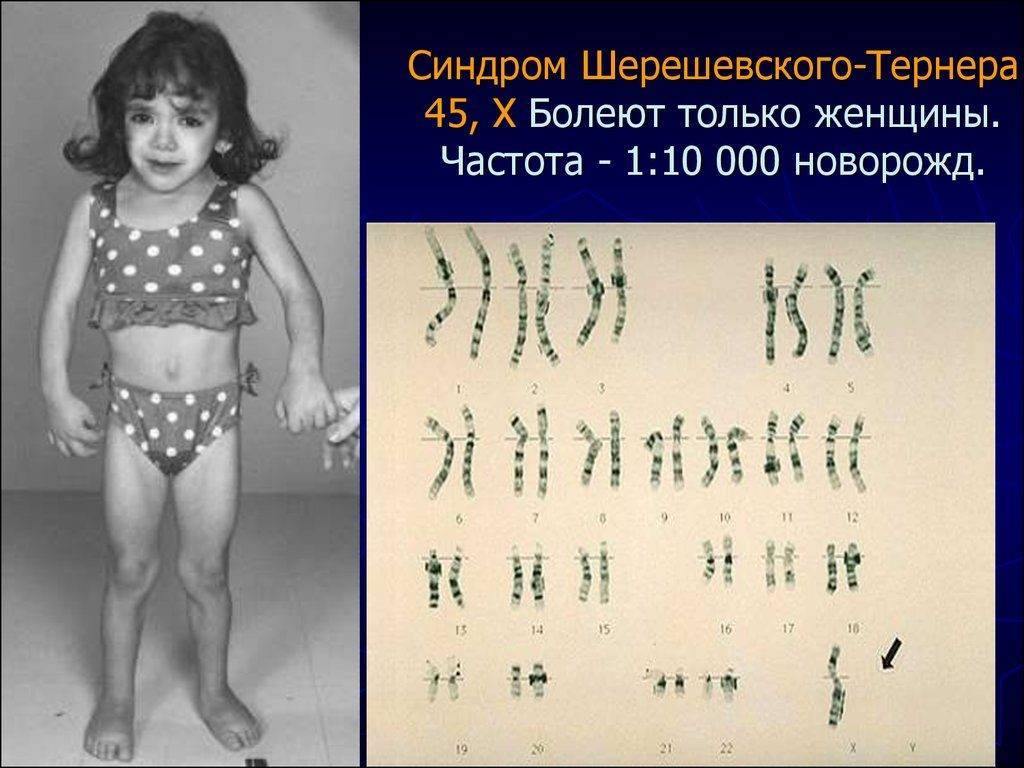


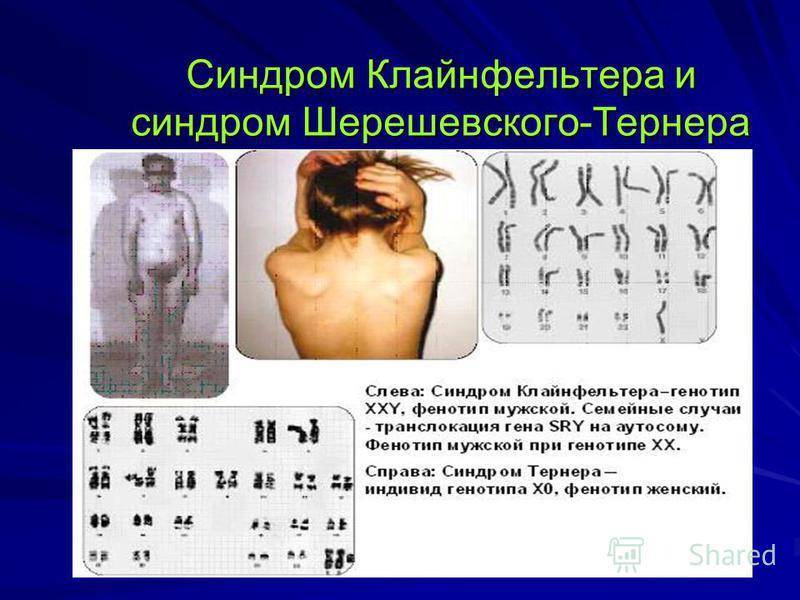





Психическая сфера больных с синдромом Шерешевского – Тернера практически не страдает. Правда, некоторые авторы отмечают снижение у них внимания и некоторых процессов восприятия, в частности пространственного, что отражается на качестве обработки невербальной (несловесной) информации.
Различают три формы дисгенезии гонад – стертая, чистая и смешанная, отличающиеся друг от друга клиническими проявлениями. При стертой форме у пациенток выявляется мозаичный кариотип 45 Х0/ 46 ХХ, и выраженность клинической картины связана с соотношением клеток с нормальным и деформированным кариотипом. Если преобладает процент клеток с отсутствием одной из Х-хромосом, то внешние признаки больного будут схожи с классическим видом пациентов с заболеванием Шерешевского – Тернера. В противном случае будет отмечаться спонтанное развитие внешних половых признаков, но у больных будет выявлена первичная аменорея и признаки недоразвития половых органов.
Чистая форма яичниковой дисгенезии (синдром Свайера) характеризуется кариотипом 46 ХХ, либо 46 ХY. По внешнему виду распознать наличие хромосомных дефектов сложно, так как рост пациенток нормальный, соматические дефекты и аномалии не выявляются. Однако, внешние половые признаки при этом слабо развиты. Кроме того, при осмотре выявляется генитальный инфантилизм, т.е. половые органы также недостаточно развиты.
Смешанная форма дисгенезии яичников проявляется первичной аменореей, вирилизацией – увеличением клитора, оволосением по мужскому типу, что обусловлено наличием в кариотипе неполноценной Y-хромосомы. При этой форме заболевания в постпубертатном периоде возможны опухоли яичников комбинированного типа – гонадобластомы, эмбриональные карциномы.